
Lipoïdose (lysosomale lipidenstapelingsziekte): wat is het, voorbeelden?
Aandacht! De informatie is alleen voor informatieve doeleinden en is niet bedoeld om een diagnose te stellen en een behandeling voor te schrijven. Raadpleeg altijd uw specialist!
Inhoud
- Wat zijn lysosomale stapelingsziekten?
- Wat zijn lipiden?
- Hoe wordt lipoidose geërfd?
- Soorten en symptomen van lysosomale stapelingsziekten
Wat zijn lysosomale stapelingsziekten?
Lysosomale stapelingsziekten (lipoïdose), zijn een groep erfelijke stofwisselingsziekten waarbij schadelijke hoeveelheden vetstoffen (lipiden) zich ophopen in verschillende cellen en weefsels van het lichaam.
Mensen met deze aandoeningen maken ofwel niet genoeg van een van de enzymen die nodig zijn om lipiden af te breken (metaboliseren), ofwel maken ze enzymen aan die niet goed werken.
Na verloop van tijd kan deze overmatige ophoping van vet leiden tot blijvende schade aan cellen en weefsels, vooral in de hersenen, het perifere zenuwstelsel (zenuwen van het ruggenmerg naar de rest van de lichaam), lever, milt en beenmerg.
Wat zijn lipiden?
Lipiden zijn vetachtige stoffen die belangrijke onderdelen zijn van de membranen in en tussen cellen en in de myelineschede, die de zenuwen bedekt en beschermt. Lipiden omvatten oliën, vetzuren, wassen, steroïden (zoals cholesterol en oestrogeen) en andere verwante verbindingen.
Deze vetstoffen worden van nature opgeslagen in de cellen, organen en weefsels van het lichaam. Kleine lichaampjes in cellen, lysosomen genaamd, zetten of metaboliseren regelmatig lipiden en eiwitten om in kleinere componenten om energie aan het lichaam te leveren. Aandoeningen waarbij intracellulair materiaal, dat niet kan worden gemetaboliseerd, in lysosomen achterblijft, worden lysosomale stapelingsziekten genoemd.
Naast ziekten die verband houden met de ophoping van lipiden, omvatten andere lysosomale ziekten die verband houden met accumulatie mucolipidosen, waarin cellen en weefsels zich ophopen. een overmatige hoeveelheid lipiden waaraan suikermoleculen zijn gehecht, evenals mucopolysaccharidosen, waarin een overmatige hoeveelheid grote, complexe suiker zich ophoopt moleculen.
Hoe worden geërfd? lipoidose?
Lysosomale stapelingsziekten worden geërfd van een of beide ouders die een defect gen dragen dat een specifiek lipide-metaboliserend enzym in een klasse cellen in het lichaam reguleert. Ze kunnen op twee manieren worden overgeërfd:
- Autosomaal recessieve overerving treedt op wanneer beide ouders een kopie van het defecte gen dragen en doorgeven, maar geen van beide ouders wordt beïnvloed door de aandoening. Elk kind van deze ouders heeft een kans van 25 procent om beide exemplaren van het defecte gen te erven, 50 procent de kans dat hij een drager zal zijn die vergelijkbaar is met zijn ouders, en een kans van 25 procent om geen kopie van de defecte te erven gen. Kinderen van beide geslachten kunnen autosomaal recessief overgeërfd worden.
- X-gebonden (of geslachtsgebonden) recessieve overerving treedt op wanneer de moeder het aangetaste gen op het X-chromosoom draagt. De X- en Y-chromosomen zijn betrokken bij geslachtsbepaling. Vrouwen hebben twee X-chromosomen, terwijl mannen één X-chromosoom en één Y-chromosoom hebben. Zonen van vrouwelijke dragers hebben 50 procent kans om de aandoening te erven en te beïnvloeden, aangezien zonen één X-chromosoom van hun moeder en één Y-chromosoom van hun vader krijgen. Dochters hebben 50 procent kans om het aangetaste X-chromosoom van hun moeder te erven en zijn drager of licht aangetast. Getroffen mannen dragen de ziekte niet over op hun zonen, maar hun dochters zullen drager en drager van de ziekte zijn.
Soorten en symptomen van lysosomale stapelingsziekten
ziekte van Gaucher - de meest voorkomende lysosomale ziekte. De ziekte wordt veroorzaakt door een tekort aan het enzym glucocerebrosidase, wat leidt tot de ophoping van glucocerebroside in veel weefsels. Vetmateriaal kan zich ophopen in de hersenen, milt, lever, nieren, longen en beenmerg.
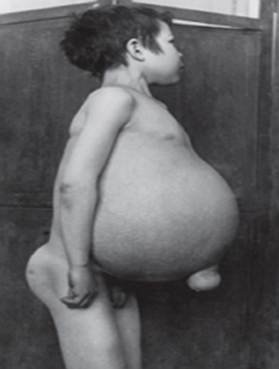
Symptomen kunnen zijn: hersenbeschadiging, vergroting van de milt en lever, verminderde leverfunctie, skeletaandoeningen en botbeschadiging die pijn en breuken kan veroorzaken, zwelling van de lymfeklieren en (soms) aangrenzende gewrichten, opgeblazen gevoel, bruinachtige huidskleur, Bloedarmoede, laag aantal bloedplaatjes en gele vlekken op de ogen.
De personen die het zwaarst getroffen zijn, kunnen ook vatbaarder zijn voor infecties. De ziekte treft mannen en vrouwen in gelijke mate.
De ziekte van Gaucher heeft drie algemene klinische subtypes:
- Type 1 (Of niet neuronopathisch type) is de meest voorkomende vorm van de ziekte in de Verenigde Staten en Europa. De hersenen worden niet aangetast, maar er kan schade zijn aan de longen en, in zeldzame gevallen, aan de nieren. Symptomen kunnen vroeg in het leven of in de volwassenheid beginnen en omvatten een vergrote lever en ernstige vergroting van de milt, wat kan leiden tot scheuren en bijkomende complicaties. Skeletzwakte en botziekte kunnen uitgebreid zijn. Mensen in deze groep krijgen meestal gemakkelijk blauwe plekken vanwege het lage aantal bloedplaatjes in hun bloed. Ze kunnen ook vermoeidheid ervaren als gevolg van bloedarmoede. Afhankelijk van het begin en de ernst van de ziekte, kunnen mensen met type 1 volwassen kan worden. Veel patiënten hebben een milde ziekte of vertonen mogelijk geen symptomen. Hoewel 1type De ziekte van Gaucher komt veel voor bij mensen van Ashkenazi-joodse afkomst en kan mensen van alle etnische achtergronden treffen.
- Typ 2 (of acute neuropathische kindertijd Ziekte van Gaucher) begint meestal binnen 3 maanden na de geboorte. Symptomen zijn onder meer uitgebreide en progressieve hersenbeschadiging, spasticiteit, toevallen, stijve ledematen, vergrote lever (lever hepatomegalie) en milt (splenomegalie), abnormale oogbewegingen en slecht zuig- en slikvermogen. Getroffen kinderen sterven meestal vóór de leeftijd van 2 jaar.
- Typ 3 (chronisch neuronopathisch vorm) kan op elk moment tijdens de kindertijd of zelfs op volwassen leeftijd beginnen. Het wordt gekenmerkt door langzaam progressieve maar minder ernstige neurologische symptomen in vergelijking met de acute ziekte van Gaucher 2 soorten. Belangrijke symptomen zijn onder meer oogbewegingsstoornissen, cognitieve stoornissen, slechte coördinatie, toevallen, vergroting van de milt en/of lever, skeletaandoeningen, bloedaandoeningen waaronder bloedarmoede, en problemen met ademen. Bijna iedereen met de ziekte van Gaucher 3 soorten, die een enzymvervangende therapie krijgt, de volwassen leeftijd bereikt.
Voor patiënten 1e en 3e type Enzymvervangingstherapie, die elke twee weken intraveneus wordt toegediend, kan de grootte van de lever en de milt drastisch verminderen, skeletafwijkingen verminderen en andere manifestaties omkeren. Een succesvolle beenmergtransplantatie behandelt de neurologische manifestaties van de ziekte. Deze procedure brengt echter aanzienlijke risico's met zich mee en wordt zelden uitgevoerd bij personen met de ziekte van Gaucher.
In zeldzame gevallen kan een operatie nodig zijn om de milt geheel of gedeeltelijk te verwijderen (als een persoon een zeer laag aantal bloedplaatjes heeft of wanneer een vergroot orgaan het comfort van de persoon sterk beïnvloedt). Bloedtransfusie kan sommige mensen met bloedarmoede ten goede komen. Anderen hebben mogelijk een gewrichtsvervangende operatie nodig om de mobiliteit en kwaliteit van leven te verbeteren. Er is momenteel geen effectieve behandeling voor de hersenbeschadiging die kan optreden bij mensen met de ziekte van Gaucher type 2 en 3.
Ziekte van Niemann-Pick is een groep autosomaal recessieve aandoeningen die wordt veroorzaakt door de ophoping van vet en cholesterol in de cellen van de lever, milt, beenmerg, longen en, in sommige gevallen, de hersenen.
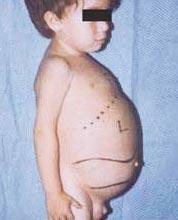
Neurologische complicaties kunnen zijn: ataxie (gebrek aan spiercoördinatie die het lopen, schrijven en eten kan beïnvloeden, onder andere functies), oogverlamming, hersendegeneratie, leerproblemen, spasticiteit, moeite met eten en slikken, onduidelijke spraak, verlies van spiertonus, overgevoeligheid voor aanraking en enige ondoorzichtigheid van het hoornvlies als gevolg van overmatige accumulatie materialen. Bij 50% van de getroffen mensen ontwikkelt zich een karakteristieke kersenrode halo rond het midden van het netvlies, die kan worden gezien door een arts met een speciaal instrument.
De ziekte van Niemann-Pick wordt ingedeeld in drie algemene subtypes:
- Type A, de meest ernstige vorm, begint in de vroege kinderjaren. Baby's zien er normaal uit bij de geboorte, maar tegen 6 maanden ontwikkelt zich diepe hersenbeschadiging, een vergrote lever en milt, en gezwollen lymfeklieren en klieren onder de huid (xanthomen). De milt kan 10 keer zo groot worden als normaal en kan scheuren, waardoor bloedingen ontstaan. Deze kinderen worden geleidelijk zwakker, verliezen motorische functies, kunnen bloedarmoede krijgen en zijn vatbaar voor terugkerende infecties. Ze leven zelden langer dan 18 maanden. Deze vorm van de ziekte komt het meest voor in Joodse families.
- Type B(of jeugdbegin) heeft meestal geen invloed op de hersenen, maar de meeste kinderen ontwikkelen zich ataxie, schade aan de zenuwen die het ruggenmerg verlaten (perifere neuropathie), en longproblemen die toenemen met de leeftijd. Een vergroting van de lever en de milt is typisch voor adolescenten. Mensen met type B kunnen relatief lang leven, maar velen hebben levenslange zuurstoftherapie nodig vanwege longschade. Niemann-Pick typen A en B zijn het resultaat van een ophoping van een vetachtige stof genaamd sfingomyeline als gevolg van een tekort aan een enzym dat sfingomyelinase wordt genoemd.
- Type C kan vroeg in het leven verschijnen of zich ontwikkelen tijdens de adolescentie of zelfs de volwassenheid. Ziekte van Niemann-Picktype C wordt niet veroorzaakt door sphlingomyelinasedeficiëntie, maar door een gebrek aan NPC1- of NPC2-eiwitten. Dientengevolge hopen verschillende lipiden en vooral cholesterol zich op in zenuwcellen en veroorzaken hun storing. De betrokkenheid van de hersenen kan uitgebreid zijn, wat resulteert in onvermogen om op en neer te kijken, moeite met lopen en slikken, progressief gehoorverlies en progressief Dementie. Mensen met type C hebben slechts een lichte vergroting van de milt en lever. Levensverwachting van mensen met type C varieert aanzienlijk. Sommige mensen sterven in de kindertijd, terwijl anderen die minder getroffen zijn, ook op volwassen leeftijd kunnen leven.
Er is momenteel geen remedie voor de ziekte van Niemann-Pick. De behandeling is ondersteunend. Kinderen sterven meestal door infectie of progressief neurologisch verlies. Er zijn gevallen geweest van beenmergtransplantatie bij verschillende patiënten met: type B met gemengde resultaten.
ziekte van Fabryook gekend als alfa-galactosidase-A-deficiëntie, veroorzaakt de ophoping van vetweefsel in het autonome zenuwstelsel (dat deel van het zenuwstelsel dat controleert onwillekeurige functies zoals ademhaling en hartslag), in de ogen, nieren en cardiovasculaire systeem.
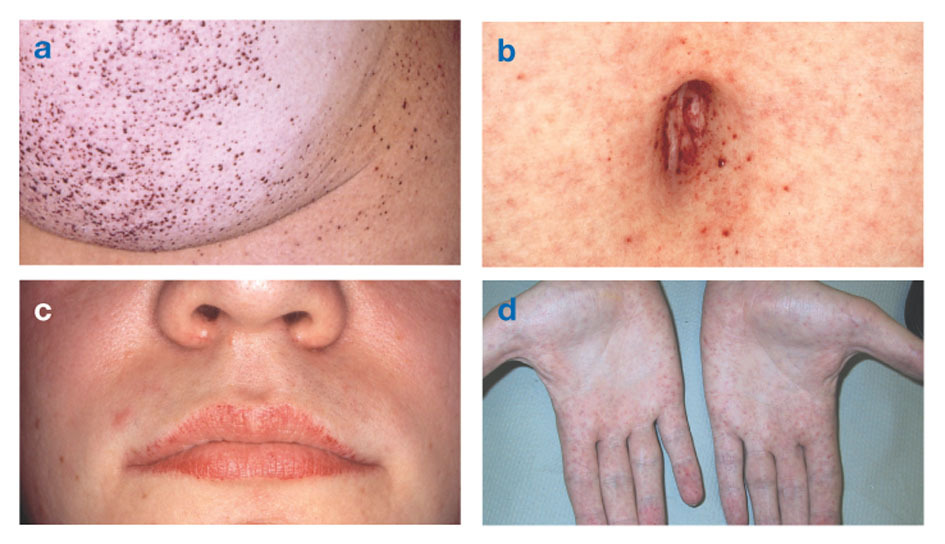
De ziekte van Fabry is de enige X-gebonden lipidenziekte. De ziekte treft vooral mannen, hoewel een mildere en meer variabele vorm bij vrouwen voorkomt. Zelden hebben getroffen vrouwen ernstige symptomen die lijken op die bij mannen met de aandoening. Het begin van de symptomen treedt meestal op tijdens de kindertijd of adolescentie.
Neurologische symptomen zijn onder meer brandende pijn in de armen en benen die erger wordt bij warm weer of daarna inspanning, evenals de ophoping van overtollig materiaal in de transparante lagen van het hoornvlies (wat leidt tot troebeling, maar zonder veranderingen in het gezichtsvermogen). Vetophoping in de wanden van bloedvaten kan de bloedsomloop verstoren, waardoor een persoon in gevaar komt hartinfarct of hartaanval. Andere symptomen zijn onder meer een vergroot hart, progressief nierfalenleidend tot nierziekte in het eindstadium, gastro-intestinale problemen, verminderd zweten en koorts.
Angiokeratomen (kleine, niet-kankerachtige, roodpaarse verheven plekken op de huid) kunnen zich in het onderlichaam ontwikkelen en komen vaker voor met de leeftijd.
Mensen met de ziekte van Fabry overlijden vaak vroegtijdig aan complicaties van: hartfalen, nierfalen of beroerte. Geneesmiddelen zoals fenytoïne en carbamazepine worden vaak voorgeschreven om de pijn die gepaard gaat met de ziekte van Fabry te behandelen, maar niet om deze te genezen. Metoclopramide of Lipisorb (voedingssupplement) kunnen maagdarmklachten verlichten die: komt vaak voor bij mensen met de ziekte van Fabry, en sommige mensen hebben mogelijk een niertransplantatie nodig of dialyse. Enzymvervanging kan de opslag verminderen, pijn verlichten en de orgaanfunctie behouden bij sommige mensen met de ziekte van Fabry.
Ziekte van Farber ook gekend als Farber's lipogranulomatose, beschrijft een groep zeldzame autosomaal recessieve aandoeningen die de ophoping van vetweefsel in gewrichten, weefsels en het centrale zenuwstelsel veroorzaken. Het treft zowel mannen als vrouwen. De ziekte begint meestal in de vroege kinderjaren, maar kan later in het leven optreden.
Kinderen met de klassieke ziekte van Farber ontwikkelen neurologische symptomen, waaronder mogelijk verhoogde slaperigheid en lethargie, en problemen met slikken. De lever, het hart en de nieren kunnen ook worden aangetast. Andere symptomen kunnen zijn: gewrichtscontracturen (chronische verkorting van spieren of pezen rond de gewrichten), braken, artritis, vergroting lymfeklieren, gewrichtsontsteking, heesheid en knobbels onder de huid die dikker worden rond de gewrichten naarmate het vordert ziekten.
Bij getroffen mensen met ademhalingsmoeilijkheden moet mogelijk een beademingsslang worden ingebracht. De meeste kinderen met deze aandoening sterven op de leeftijd van 2, meestal door: longziekten. Bij een van de meest ernstige vormen van de ziekte kunnen kort na de geboorte een vergrote lever en milt worden vastgesteld. Baby's die met deze vorm van de ziekte worden geboren, sterven meestal binnen 6 maanden.
De ziekte van Farber wordt veroorzaakt door een tekort aan een enzym dat ceramidase wordt genoemd. Er is momenteel geen specifieke behandeling voor de ziekte van Farber. Corticosteroïden kunnen worden voorgeschreven om pijn te verlichten. Beenmergtransplantatie kan granulomen (kleine massa's ontstoken weefsel) verminderen bij mensen met een milde ontsteking of bij patiënten zonder complicaties aan de longen of het zenuwstelsel. Bij oudere mensen kunnen granulomen worden verkleind of operatief worden verwijderd.
gangliosidose bestaat uit twee verschillende groepen genetische ziekten. Beide zijn autosomaal recessief en beïnvloeden zowel mannen als vrouwen.
GM1 gangliosidose.
GM1-gangliosidose wordt veroorzaakt door een tekort aan het enzym bèta-galactosidase, wat resulteert in een abnormale accumulatie van zure lipidenmaterialen, met name in zenuwcellen in het centrale en perifere zenuwstelsel systemen. GM1-gangliosidose heeft drie klinische manifestaties:
- GM1 (het meest ernstige subtype, dat snel na de geboorte optreedt) kan neurodegeneratie, toevallen, vergrote lever en milt, ruwheid van gelaatstrekken, skeletonregelmatigheden, gewrichtsstijfheid, opgeblazen gevoel, spierzwakte, overdreven schrikreactie en problemen met gang. Ongeveer de helft van de getroffenen ontwikkelt kersenrode vlekken in het oog. Kinderen kunnen op de leeftijd van 1 doof en blind zijn en sterven vaak op de leeftijd van 3 aan hartcomplicaties of longontsteking.
- laat kind GM1-gangliosidose begint meestal tussen de leeftijd van 1 en 3 jaar. Neurologische symptomen zijn onder meer ataxie, toevallen, dementie en moeite met spreken.
- GM1-gangliosidose ontwikkelt zich tussen de 3 en 30 jaar. Symptomen zijn onder meer verminderde spiermassa (spieratrofie), neurologische complicaties die minder ernstig zijn en langzamer vorderen dan andere vormen aandoeningen, troebelheid van het hoornvlies bij sommige mensen en aanhoudende spiersamentrekkingen die draaiende en repetitieve bewegingen of abnormale houdingen veroorzaken (dystonie). Angiokeratomen kunnen zich ontwikkelen in de onderste romp van het lichaam. De grootte van de lever en milt is normaal bij de meeste getroffen mensen.
GM2 gangliosidose.
GM2-gangliosidose zorgt er ook voor dat het lichaam overtollige zure vetstoffen ophoopt in weefsels en cellen, voornamelijk zenuwcellen. Deze aandoeningen zijn het gevolg van een tekort aan het enzym beta-hexosaminidase. GM2-aandoeningen zijn onder meer:
- Tay-Sachs ziekte (ook bekend als GM2-gangliosidose variant B) en zijn variante vormen worden veroorzaakt door een tekort aan het enzym hexosaminidase A. Getroffen kinderen ontwikkelen zich normaal tijdens de eerste paar maanden van hun leven. Symptomen beginnen op de leeftijd van 6 maanden en omvatten progressief verlies van mentale capaciteit, dementie, verminderd oogcontact, toegenomen een schrikreactie op geluid, progressief gehoorverlies leidend tot doofheid, moeite met slikken, blindheid, kersenrode vlekken op het netvlies en sommige verlamming. Epileptische aanvallen kunnen beginnen in het tweede levensjaar van een kind. Baby's kunnen uiteindelijk afhankelijk worden van de voedingssonde en sterven vaak op 4-jarige leeftijd aan terugkerende infecties. Er is helaas geen speciale behandeling. Anticonvulsiva kunnen aanvankelijk aanvallen beheersen. Andere ondersteunende behandelingen zijn onder meer goede voeding en hydratatie en methoden om de luchtwegen open te houden. Een zeldzame vorm van de aandoening genaamd laat optredende ziekte van Tay-Sachs, komt voor bij mensen tussen de 20 en 30 jaar en wordt gekenmerkt door een onstabiele gang en progressieve neurologische achteruitgang.
- ziekte van Sandhoff (Optie AB) is een ernstige vorm van de ziekte van Tay-Sachs. Het begint meestal op de leeftijd van 6 maanden en is niet beperkt tot een etnische groep. Neurologische symptomen kunnen zijn: progressieve verslechtering van het centrale zenuwstelsel, motorische zwakte, vroege blindheid, ernstige angstreactie op geluid, spasticiteit, shock of spiertrekkingen spieren (myoclonus), epilepsie, een abnormaal vergroot hoofd (macrocefalie) en rode vlekken in het oog. Andere symptomen kunnen zijn: frequente luchtweginfecties, hartruis, popachtige gelaatstrekken en vergroting van de lever en milt. Er is geen specifieke behandeling voor de ziekte van Sandhoff. Net als bij de ziekte van Tay-Sachs, omvat ondersteunende zorg het te allen tijde openhouden van de luchtwegen, evenals goede voeding en hydratatie. Anticonvulsiva kunnen aanvankelijk epilepsie (aanvallen) onder controle houden.
ziekte van Krabbe(ook gekend als globulaire cel leukodystrofie en galactosylceramide lipidose) is een autosomaal recessieve ziekte die wordt veroorzaakt door een tekort aan het enzym galactocerebrosidase. De ziekte treft meestal zuigelingen met het begin vóór de leeftijd van 6 maanden, maar kan optreden tijdens de adolescentie of volwassenheid.
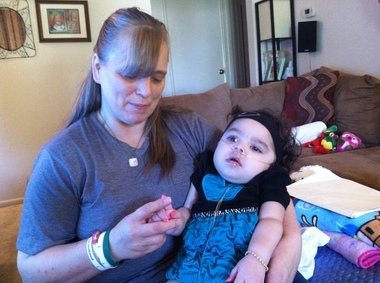
De ophoping van onverteerde vetten beïnvloedt de groei van de beschermende omhulling van de zenuw (myelineschede) en veroorzaakt ernstige aantasting van mentale en motorische vaardigheden. Andere symptomen zijn spierzwakte, verminderd rekvermogen van de spieren (hypotonie), spierspanning (spasticiteit), plotselinge shock of spiertrekkingen van de ledematen (myoclonische aanvallen), prikkelbaarheid, onverklaarbare koorts, doofheid, blindheid, verlamming en slikproblemen. Ook kan er langdurig gewichtsverlies optreden.
De ziekte kan worden gediagnosticeerd door middel van enzymtesten en het identificeren van de karakteristieke celgroepering in bolvormige lichamen in de witte stof van de hersenen, demyelinisatie van zenuwen en degeneratie, en vernietiging van hersencellen. Bij zuigelingen is de ziekte meestal fataal vóór de leeftijd van 2 jaar. Mensen met een meer gevorderde vorm van de ziekte hebben een milder verloop van de ziekte en leven aanzienlijk langer.
Er is geen specifieke behandeling voor de ziekte van Krabbe ontwikkeld, hoewel vroege beenmergtransplantatie sommige mensen kan helpen. Mensen met een meer gevorderde vorm van de ziekte hebben een milder verloop van de ziekte en leven aanzienlijk langer.
Metachromatische leukodystrofie, of MLD, is een groep aandoeningen die wordt gekenmerkt door de ophoping van vetstoffen in de witte stof van het centrale zenuwstelsel en in perifere zenuwen en tot op zekere hoogte in de nieren. Net als bij de ziekte van Krabbe beïnvloedt MLD de myeline, die de zenuwen bedekt en beschermt. Deze autosomaal recessieve aandoening wordt veroorzaakt door een tekort aan het enzym arylsulfatase A. Deze aandoening treft zowel mannen als vrouwen.
Metachromatische leukodystrofie heeft drie karakteristieke vormen: late baby, juveniele en volwassen.
- Late baby MLD begint meestal tussen 12 en 20 maanden na de geboorte. Kinderen kunnen in het begin normaal lijken, maar hebben moeite met lopen en hebben de neiging om te vallen, vergezeld van af en toe pijn in de armen en benen. progressief verlies van gezichtsvermogen leidend tot blindheid, ontwikkelingsachterstanden en verlies van eerder verworven mijlpalen, verminderd slikken, toevallen en dementie tot 2 jaar. Kinderen ontwikkelen ook progressieve spierafbraak en zwakte en verliezen uiteindelijk hun vermogen om te lopen. De meeste kinderen met deze vorm van de aandoening sterven op de leeftijd van 5 jaar.
- Juveniele MLD begint meestal tussen de leeftijd van 3 en 10 jaar. Symptomen zijn onder meer verminderde schoolprestaties, mentale stoornissen, ataxie, toevallen en dementie. Symptomen nemen toe, waarbij de dood 10-20 jaar na het begin van de ziekte optreedt.
- Volwassen vorm. Symptomen volwassenen begint na de leeftijd van 16 jaar en kan bestaan uit ataxie, toevallen, abnormale tremoren van de ledematen (tremoren), verminderde concentratie, depressie, psychische stoornissen en dementie. De dood treedt meestal 6-14 jaar na het begin van de symptomen op.
MLD is niet te genezen. De behandeling is symptomatisch en ondersteunend. Beenmergtransplantatie kan in sommige gevallen de progressie van de ziekte vertragen. Er is aanzienlijke vooruitgang geboekt met gentherapie in diermodellen met MLD en in klinische onderzoeken.
ziekte van Wolmanook gekend als lysosomale zure lipasedeficiëntieis een ernstige stoornis in de vetopslag die gewoonlijk fataal is op de leeftijd van 1 jaar. Deze autosomaal recessieve aandoening wordt gekenmerkt door een ophoping van cholesterolesters (meestal de transportvorm van cholesterol) en triglyceriden (de chemische vorm waarin vetten in het lichaam voorkomen), die zich aanzienlijk kunnen ophopen en celbeschadiging kunnen veroorzaken en stoffen.
Zowel mannen als vrouwen lijden aan deze aandoening. Zuigelingen zijn normaal en actief bij de geboorte, maar ontwikkelen snel progressieve mentale stoornissen, een vergrote lever en sterk vergrote milt, een opgeblazen gevoel, gastro-intestinale problemen. geelzucht, bloedarmoede, braken en kalkaanslag in bijnierenwaardoor ze hard worden.
een ander type lysosomale zure lipasedeficiëntie is cholesterolesterstapelingsziekte. Deze uiterst zeldzame ziekte treedt op als gevolg van de opslag van cholesterolesters en triglyceriden in bloedcellen, lymfe en lymfoïde weefsel. Bij kinderen op volwassen leeftijd wordt de lever groter, wat leidt tot cirrose en chronisch Leverfalen. Kinderen kunnen ook calciumafzettingen in de bijnieren hebben en geelzucht kan zich in de latere stadia van de ziekte ontwikkelen.
Momenteel wordt de kwestie van enzymvervanging actief bestudeerd, zowel bij de ziekte van Wolman als bij de accumulatie van een cholesterolester.
Aandacht! De informatie is alleen voor informatieve doeleinden en is niet bedoeld om een diagnose te stellen en een behandeling voor te schrijven. Raadpleeg altijd uw specialist!


