
Lipoidoza (lizosomska bolezen shranjevanja lipidov): kaj je to, primeri
Pozor! Informacije so zgolj informativne narave in niso namenjene diagnosticiranju in predpisovanju zdravljenja. Vedno se posvetujte s svojim zdravnikom specialistom!
Vsebina
- Kaj so lizosomske bolezni shranjevanja?
- Kaj so lipidi?
- Kako se lipoidoza podeduje?
- Vrste in simptomi lizosomskih bolezni shranjevanja
Kaj so lizosomske bolezni shranjevanja?
Lizosomske bolezni shranjevanja (lipoidoza), so skupina dednih presnovnih bolezni, pri katerih se v različnih celicah in tkivih telesa kopičijo škodljive količine maščobnih snovi (lipidov).
Ljudje s temi motnjami bodisi ne proizvedejo dovolj enega od encimov, potrebnih za razgradnjo (presnovo) lipidov, ali pa proizvajajo encime, ki ne delujejo pravilno.
Sčasoma lahko to prekomerno kopičenje maščobe povzroči trajne poškodbe celic in tkiv, zlasti v možganih, perifernem živčnem sistemu (živci od hrbtenjače do preostalega telo), jetra, vranica in kostnega mozga.
Kaj so lipidi?
Lipidi so maščobi podobne snovi, ki so pomembni deli membran znotraj in med celicami ter v mielinski ovojnici, ki pokriva in ščiti živce. Lipidi vključujejo olja, maščobne kisline, voske, steroide (kot sta holesterol in estrogen) in druge sorodne spojine.
Te maščobne snovi so naravno shranjene v celicah, organih in tkivih telesa. Majhna telesa v celicah, imenovana lizosomi, redno pretvarjajo ali presnavljajo lipide in beljakovine v manjše komponente, da telesu zagotovijo energijo. Motnje, pri katerih znotrajcelični material, ki ga ni mogoče presnoviti, ostane v lizosomih, imenujemo lizosomske bolezni shranjevanja.
Poleg bolezni, povezanih s kopičenjem lipidov, druge lizosomske bolezni, povezane z kopičenjem, vključujejo mukolipidoze, pri katerih se kopičijo celice in tkiva. prekomerna količina lipidov z vezanimi molekulami sladkorja, pa tudi mukopolisaharidoze, pri katerih se kopiči prevelika količina velikega kompleksnega sladkorja molekule.
Kako so podedovani lipoidoza?
Lizosomske bolezni shranjevanja so podedovane od enega ali obeh staršev, ki nosita okvarjen gen, ki uravnava specifičen encim za presnovo lipidov v razredu celic v telesu. Lahko se podedujejo na dva načina:
- Avtosomno recesivno dedovanje se pojavi, ko oba starša nosita in prenašata kopijo okvarjenega gena, vendar motnja ni prizadeta nobenega od staršev. Vsak otrok, rojen pri teh starših, ima 25-odstotno možnost, da podeduje obe kopiji okvarjenega gena, 50-odstotno verjetnost, da bo nosilec podoben svojim staršem, in 25-odstotna možnost, da ne bo podedoval nobene kopije okvarjenega gen. Otroci obeh spolov se lahko podedujejo avtosomno recesivno.
- X-vezano (ali spolno povezano) recesivno dedovanje se pojavi, ko mati nosi prizadeti gen na kromosomu X. Kromosoma X in Y sodelujeta pri določanju spola. Ženske imajo dva kromosoma X, moški pa en kromosom X in en kromosom Y. Sinovi nosilk imajo 50-odstotno možnost, da podedujejo in prizadenejo motnjo, saj sinovi prejmejo en kromosom X od matere in en kromosom Y od očeta. Hčerke imajo 50-odstotno možnost, da podedujejo prizadeti kromosom X po materi in so nosilke ali blago prizadete. Prizadeti moški ne prenašajo bolezni na svoje sinove, bodo pa njihove hčere nosilke in nosilke bolezni.
Vrste in simptomi lizosomskih bolezni shranjevanja
Gaucherjeva bolezen - najpogostejša med lizosomskimi boleznimi. Bolezen je posledica pomanjkanja encima glukocerebrozidaze, kar vodi do kopičenja glukocerebrozida v številnih tkivih. Maščobna snov se lahko kopiči v možganih, vranici, jetrih, ledvicah, pljučih in kostnem mozgu.
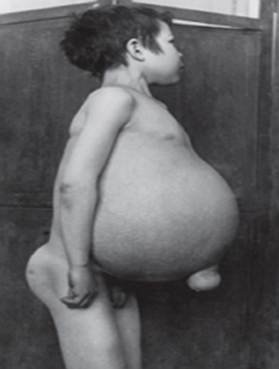
Simptomi lahko vključujejo poškodbe možganov, povečanje vranice in jetra, okvarjeno delovanje jeter, skeletne bolezni in poškodbe kosti, ki lahko povzročijo bolečine in zlome, otekanje bezgavk in (včasih) sosednjih sklepov, napihnjenost, rjavkast ten kože, anemija, nizko število trombocitov in rumene lise na očeh.
Posamezniki, ki so najbolj prizadeti, so lahko tudi bolj dovzetni za okužbe. Bolezen enako prizadene moške in ženske.
Gaucherjeva bolezen ima tri splošne klinične podtipe:
- Vrsta 1 (Ali ne nevronopatsko tip) je najpogostejša oblika bolezni v Združenih državah in Evropi. Možgani niso prizadeti, lahko pa pride do poškodb pljuč in v redkih primerih ledvic. Simptomi se lahko začnejo zgodaj v življenju ali v odrasli dobi in vključujejo povečana jetra in močno povečanje vranice, kar lahko povzroči rupturo in dodatne zaplete. Slabost skeleta in bolezni kosti sta lahko obsežni. Ljudje v tej skupini običajno zlahka dobijo modrice zaradi nizkega števila trombocitov v krvi. Prav tako lahko občutijo utrujenost zaradi anemije. Odvisno od začetka in resnosti bolezni so ljudje s vrsta 1 lahko doživi odraslost. Mnogi bolniki imajo blago bolezen ali morda ne kažejo nobenih simptomov. Čeprav 1tip Gaucherjeva bolezen je pogosta med ljudmi aškenazijske judovske dediščine in lahko prizadene ljudi vseh etničnih okolij.
- Vrsta 2 (oz akutna otroška nevropatija Gaucherjeva bolezen) se običajno začne v 3 mesecih po rojstvu. Simptomi vključujejo obsežne in progresivne poškodbe možganov, spastičnost, epileptične napade, otrdelost okončin, povečana jetra (jetrna hepatomegalija) in vranico (splenomegalija), nenormalno gibanje oči in slabo sposobnost sesanja in požiranja. Prizadeti otroci običajno umrejo pred 2 leti.
- Vrsta 3 (kronična nevronopatska oblika) se lahko začne kadar koli v otroštvu ali celo v odrasli dobi. Zanj so značilni počasi napredujoči, a manj hudi nevrološki simptomi v primerjavi z akutno Gaucherjevo boleznijo 2 vrsti. Glavni simptomi vključujejo motnje gibanja oči, kognitivne pomanjkljivosti, slabo koordinacijo, epileptične napade, povečanje vranice in/ali jeter, bolezni okostja, krvne bolezni, vključno z anemijo, in težave z dihanje. Skoraj vsi z Gaucherjevo boleznijo 3 vrste, ki prejema encimsko nadomestno zdravljenje, bo dosegel odraslo dobo.
Za bolnike 1. in 3. vrste Encimsko nadomestno zdravljenje, ki se daje intravensko vsaka dva tedna, lahko dramatično zmanjša velikost jeter in vranice, zmanjša nepravilnosti skeleta in obrne druge manifestacije. Uspešna presaditev kostnega mozga zdravi nevrološke manifestacije bolezni. Vendar pa ta postopek prinaša velika tveganja in se redko izvaja pri posameznikih z Gaucherjevo boleznijo.
V redkih primerih bo morda potrebna operacija za odstranitev celotne ali dela vranice (če ima oseba zelo nizko število trombocitov ali če povečan organ močno vpliva na udobje osebe). Transfuzija krvi lahko koristi nekaterim ljudem z anemijo. Drugi bodo morda potrebovali operacijo zamenjave sklepov za izboljšanje mobilnosti in kakovosti življenja. Trenutno ni učinkovitega zdravljenja za poškodbe možganov, ki se lahko pojavijo pri ljudeh z Gaucherjevo boleznijo tipa 2 in 3.
Niemann-Pickova bolezen je skupina avtosomno recesivnih motenj, ki nastanejo zaradi kopičenja maščob in holesterola v celicah jeter, vranice, kostnega mozga, pljuč in v nekaterih primerih možganov.
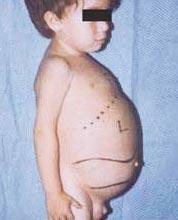
Nevrološki zapleti lahko vključujejo ataksijo (pomanjkanje mišične koordinacije, ki lahko vpliva na hojo, pisanje in prehranjevanje, med drugimi funkcijami), paraliza oči, degeneracija možganov, težave z učenjem, spastičnost, težave pri hranjenju in požiranju, nejasno govor, izguba mišičnega tonusa, preobčutljivost na dotik in nekaj motnosti roženice zaradi prekomernega kopičenja materiali. Pri 50 % obolelih se okoli središča mrežnice razvije značilen češnjevordeči halo, ki ga lahko opazi zdravnik s posebnim instrumentom.
Niemann-Pickova bolezen je razvrščena v tri splošne podtipe:
- Vrsta A, najtežja oblika, se začne v zgodnjem otroštvu. Otroci so ob rojstvu videti normalni, vendar se pri 6 mesecih razvijejo globoke poškodbe možganov, povečana jetra in vranica ter otekle bezgavke in vozlišča pod kožo (ksantomi). Vranica lahko postane 10-krat večja od običajne velikosti in lahko poči, kar povzroči krvavitev. Ti otroci postajajo progresivno šibkejši, izgubijo motorično funkcijo, lahko postanejo anemični in so nagnjeni k ponavljajočim se okužbam. Redko živijo več kot 18 mesecev. Ta oblika bolezni je najpogostejša v judovskih družinah.
- Vrsta B(ali juvenilni začetek) običajno ne vpliva na možgane, vendar se večina otrok razvije ataksijapoškodbe živcev, ki zapuščajo hrbtenjačo (periferna nevropatija), in pljučne težave, ki napredujejo s starostjo. Povečanje jeter in vranice je značilno za mladostnike. Ljudje z tip B lahko živi relativno dolgo, vendar mnogi zaradi poškodb pljuč potrebujejo doživljenjsko terapijo s kisikom. Niemann-Pick tipa A in B sta posledica kopičenja maščobne snovi, imenovane sfingomielin, zaradi pomanjkanja encima, imenovanega sfingomielinaza.
- Tip C se lahko pojavi zgodaj v življenju ali se razvije v adolescenci ali celo odrasli dobi. Niemann-Pickova bolezentip C ni posledica pomanjkanja sflingomijelinaze, temveč pomanjkanja proteinov NPC1 ali NPC2. Posledično se v živčnih celicah kopičijo različni lipidi in predvsem holesterol, ki povzročajo njihovo okvaro. Vpletenost možganov je lahko obsežna, kar povzroči nezmožnost pogleda navzgor in navzdol, težave pri hoji in požiranju, progresivno izgubo sluha in progresivno demenca. Ljudje z tip C imajo le blago povečanje vranice in jeter. Pričakovana življenjska doba ljudi s tip C se precej razlikuje. Nekateri ljudje umrejo v otroštvu, drugi, ki so manj prizadeti, pa lahko živijo tudi v odrasli dobi.
Trenutno ni zdravila za Niemann-Pickovo bolezen. Zdravljenje je podporno. Otroci običajno umrejo zaradi okužbe ali progresivne nevrološke izgube. Obstajajo primeri presaditve kostnega mozga pri več bolnikih z tip B z mešanimi rezultati.
Fabryjeva bolezenpoznan tudi kot pomanjkanje alfa-galaktozidaze-A, povzroča kopičenje maščobe v avtonomnem živčnem sistemu (tistem delu živčnega sistema, ki nadzoruje nehotene funkcije, kot sta dihanje in srčni utrip), v očeh, ledvicah in srčno-žilnem sistem.
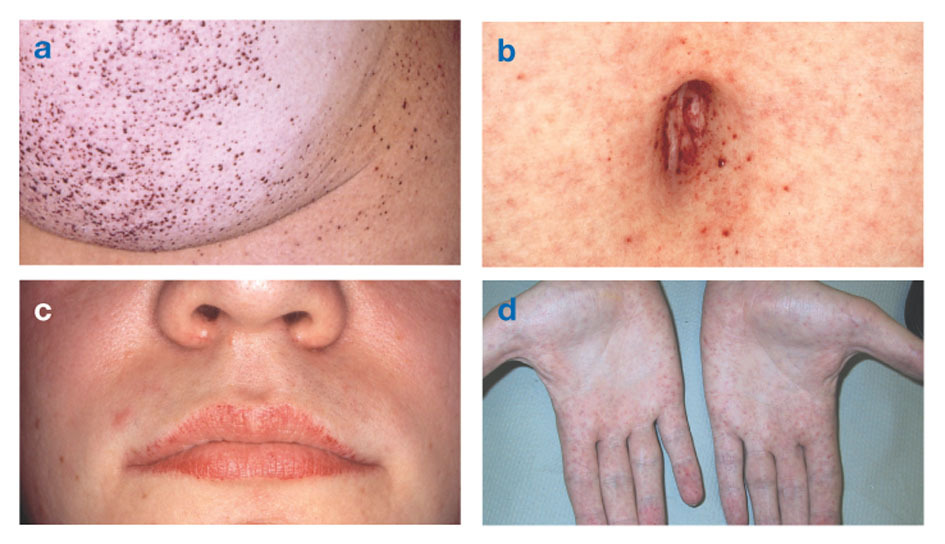
Fabryjeva bolezen je edina bolezen lipidov, vezanih na X. Bolezen prizadene predvsem moške, čeprav se pri ženskah pojavlja blažja in bolj spremenljiva oblika. Redko imajo prizadete ženske hude simptome, podobne tistim pri moških z motnjo. Simptomi se običajno pojavijo v otroštvu ali adolescenci.
Nevrološki znaki vključujejo pekoče bolečine v rokah in nogah, ki se poslabšajo v vročem vremenu ali po njem vadbo, kot tudi kopičenje odvečnega materiala v prozornih plasteh roženice (kar vodi do motnosti, vendar brez spremembe vida). Kopičenje maščobe v stenah krvnih žil lahko moti cirkulacijo in ogroža osebo možganska kap oz srčni napad. Drugi simptomi vključujejo povečano srce, progresivno odpoved ledvickar vodi v končno ledvično odpoved, težave s prebavili, zmanjšano potenje in zvišano telesno temperaturo.
Angiokeratomi (majhne, nekancerozne, rdečkasto-vijolične dvignjene lise na koži) se lahko razvijejo v spodnjem delu trupa in s starostjo postanejo pogostejši.
Ljudje s Fabryjevo boleznijo pogosto umrejo predčasno zaradi zapletov odpoved srca, odpoved ledvic ali možganska kap. Zdravila, kot sta fenitoin in karbamazepin, se pogosto predpisujejo za zdravljenje bolečine, ki spremlja Fabryjevo bolezen, ne pa za njeno ozdravitev. Metoklopramid ali Lipisorb (prehransko dopolnilo) lahko lajšata prebavne motnje se pogosto pojavlja pri ljudeh s Fabryjevo boleznijo, nekateri pa bodo morda potrebovali presaditev ledvice oz dializa. Encimska zamenjava lahko zmanjša shranjevanje, lajša bolečine in ohranja delovanje organov pri nekaterih ljudeh s Fabryjevo boleznijo.
Farberjeva bolezen poznan tudi kot Farberjeva lipogranulomatoza, opisuje skupino redkih avtosomno recesivnih motenj, ki povzročajo kopičenje maščobnih snovi v sklepih, tkivih in centralnem živčnem sistemu. Prizadene tako moške kot ženske. Bolezen se običajno začne v zgodnjem otroštvu, lahko pa se pojavi tudi kasneje v življenju.
Razvijajo se otroci s klasično Farberjevo boleznijo nevrološki simptomi, ki lahko vključujejo povečano zaspanost in letargijo ter težave z požiranje. Prizadeta so lahko tudi jetra, srce in ledvice. Drugi simptomi lahko vključujejo sklepne kontrakture (kronično skrajšanje mišic ali kit okoli sklepov), bruhanje, artritis, povečanje bezgavke, vnetje sklepov, hripavost in grudice pod kožo, ki se med napredovanjem zgostijo okoli sklepov bolezni.
Prizadeti ljudje s težavami z dihanjem bodo morda morali vstaviti dihalno cev. Večina otrok s tem stanjem umre do 2. leta starosti, običajno zaradi pljučne bolezni. Pri eni najtežjih oblik bolezni lahko kmalu po rojstvu diagnosticiramo povečana jetra in vranico. Dojenčki, rojeni s to obliko bolezni, običajno umrejo v 6 mesecih.
Farberjevo bolezen povzroča pomanjkanje encima, imenovanega ceramidaza. Trenutno ni posebnega zdravljenja za Farberjevo bolezen. Za lajšanje bolečin se lahko predpišejo kortikosteroidi. Presaditev kostnega mozga lahko zmanjša granulome (majhne mase vnetega tkiva) pri ljudeh z blagim vnetjem ali pri bolnikih brez zapletov pljučnega ali živčnega sistema. Pri starejših ljudeh je mogoče granulome zmanjšati ali kirurško odstraniti.
Gangliozidoza sestavljata dve različni skupini genetskih bolezni. Oba sta avtosomno recesivna in enako prizadeneta moške in ženske.
GM1 gangliozidoza.
Gangliozidoza GM1 je posledica pomanjkanja encima beta-galaktozidaze, kar povzroči nenormalno kopičenje kisle lipidne snovi, zlasti v živčnih celicah v osrednjem in perifernem živčevju sistemov. Gangliozidoza GM1 ima tri klinične manifestacije:
- GM1 (najhujši podtip, ki se pojavi kmalu po rojstvu) lahko vključuje nevrodegeneracijo, epileptične napade, povečana jetra in vranico, grobost obraznih potez, nepravilnosti skeleta, togost sklepov, napihnjenost, mišična oslabelost, pretiran odziv na prestrašenost in težave z hoja. Pri približno polovici prizadetih se v očesu pojavijo češnjevo rdeče lise. Otroci so lahko do 1. leta gluhi in slepi in pogosto do 3. leta starosti umrejo zaradi srčnih zapletov oz pljučnica.
- Pozni otrok Gangliozidoza GM1 se običajno začne med 1. in 3. letom starosti. Nevrološki simptomi vključujejo ataksijo, epileptične napade, demenco in težave pri govoru.
- GM1 gangliozidoza se razvije v starosti od 3 do 30 let. Simptomi vključujejo zmanjšano mišično maso (atrofijo mišic), nevrološke zaplete, ki so manj resni in napredujejo počasneje kot druge oblike motnje, motnost roženice pri nekaterih ljudeh in trajne mišične kontrakcije, ki povzročajo zvijanje in ponavljajoče se gibe ali nenormalne drže (distonija). Angiokeratomi se lahko razvijejo v spodnjem delu telesa. Velikost jeter in vranice je pri večini prizadetih ljudi normalna.
GM2 gangliozidoza.
Gangliozidoza GM2 povzroči tudi kopičenje v telesu odvečnih kislih maščobnih snovi v tkivih in celicah, predvsem v živčnih celicah. Te motnje so posledica pomanjkanja encima beta-heksosaminidaze. Motnje GM2 vključujejo:
- Tay-Sachsova bolezen (znana tudi kot različica gangliozidoze GM2 B) in njene različice so posledica pomanjkanja encima heksozaminidaze A. Prizadeti otroci se v prvih nekaj mesecih življenja normalno razvijajo. Simptomi se pojavijo pri 6 mesecih starosti in vključujejo progresivno izgubo duševne sposobnosti, demenco, zmanjšan očesni stik, povečano presenetljiv odziv na hrup, progresivna izguba sluha, ki vodi v gluhost, težave pri požiranju, slepoto, češnjevo rdeče lise na mrežnici in nekatere paraliza. Napadi se lahko začnejo v drugem letu otrokovega življenja. Dojenčki lahko sčasoma postanejo odvisni od cevke za hranjenje in pogosto umrejo do 4. leta starosti zaradi ponavljajočih se okužb. Žal posebnega zdravljenja ni. Antikonvulzivi lahko sprva nadzorujejo epileptične napade. Druga podporna zdravljenja vključujejo pravilno prehrano in hidracijo ter metode za ohranjanje odprtih dihalnih poti. Redka oblika motnje, imenovana pozno nastopajoče Tay-Sachsove bolezni, se pojavlja pri ljudeh, starih od 20 do 30 let, zanj pa je značilna nestabilna hoja in progresivno nevrološko poslabšanje.
- Sandhoffova bolezen (Možnost AB) je huda oblika Tay-Sachsove bolezni. Začetek se običajno pojavi pri starosti 6 mesecev in ni omejen na nobeno etnično skupino. Nevrološki znaki lahko vključujejo progresivno poslabšanje centralnega živčnega sistema, motorike šibkost, zgodnja slepota, huda prestrašena reakcija na zvok, spastičnost, šok ali trzanje mišice (mioklonus), epilepsija, nenormalno povečana glava (makrocefalija) in rdeče lise v očesu. Drugi simptomi lahko vključujejo pogoste okužbe dihal, šum na srcu, lutkam podobne poteze obraza ter povečanje jeter in vranice. Za Sandhoffovo bolezen ni posebnega zdravljenja. Tako kot pri Tay-Sachsovi bolezni tudi podporna oskrba vključuje ohranjanje odprtih dihalnih poti ves čas, pa tudi pravilno prehrano in hidracijo. Antikonvulzivi lahko na začetku nadzorujejo epilepsijo (krče).
Krabbejeva bolezen(poznan tudi kot globularna celična levkodistrofija in galaktozilceramidna lipidoza) je avtosomno recesivna bolezen, ki nastane zaradi pomanjkanja encima galaktocerebrozidaze. Bolezen najpogosteje prizadene dojenčke z začetkom pred 6. mesecem starosti, lahko pa se pojavi v adolescenci ali odrasli dobi.
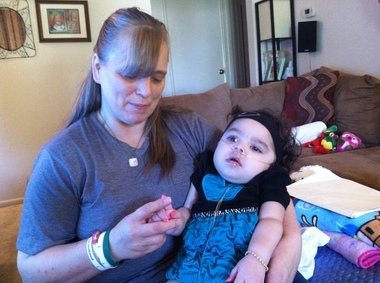
Kopičenje neprebavljenih maščob vpliva na rast zaščitne ovojnice živca (mielinske ovojnice) in povzroča hude motnje umskih in motoričnih sposobnosti. Drugi simptomi vključujejo mišično oslabelost, zmanjšano sposobnost raztezanja mišic (hipotonija), mišično napetost (spastičnost), nenaden šok ali trzanje okončin (mioklonični napadi), razdražljivost, nepojasnjena vročina, gluhost, slepota, paraliza in težave pri požiranju. Lahko pride tudi do dolgotrajne izgube teže.
Bolezen je mogoče diagnosticirati s testiranjem encimov in določitvijo njene značilne skupine celic v globoidna telesa v beli snovi možganov, demielinizacijo živcev in degeneracijo ter uničenje možganskih celic. Pri dojenčkih je bolezen običajno usodna pred 2. letom starosti. Ljudje z naprednejšo obliko bolezni imajo blažji potek bolezni in živijo bistveno dlje.
Specifičnega zdravljenja za Krabbejevo bolezen ni bilo razvito, čeprav bi nekaterim ljudem lahko pomagala zgodnja presaditev kostnega mozga. Ljudje z naprednejšo obliko bolezni imajo blažji potek bolezni in živijo bistveno dlje.
Metakromatska levkodistrofija, ali MLD, je skupina motenj, ki jih zaznamuje kopičenje maščobnih snovi v beli snovi centralnega živčnega sistema in v perifernih živcih ter do neke mere v ledvicah. Podobno kot pri Krabbejevi bolezni tudi MLD vpliva na mielin, ki pokriva in ščiti živce. To avtosomno recesivno motnjo povzroča pomanjkanje encima arilsulfataze A. Ta motnja prizadene tako moške kot ženske.
Metakromatska levkodistrofija ima tri značilne oblike: pozno dojenčke, mladostnike in odrasle.
- MLD pozno dojenčkov običajno se začne med 12 in 20 meseci po rojstvu. Otroci so lahko sprva videti normalni, vendar imajo težave s hojo in nagnjenost k padcem, ki jih spremljajo občasne bolečine v rokah in nogah. progresivna izguba vida, ki vodi v slepoto, razvojne zamude in izgubo predhodno pridobljenih mejnikov, okvarjeno požiranje, epileptične napade in demenco do 2 let. Otroci razvijejo tudi progresivno izgubljanje mišic in šibkost ter sčasoma izgubijo sposobnost hoje. Večina otrok s to obliko motnje umre do 5. leta starosti.
- Mladoletni MLD običajno se začne v starosti od 3 do 10 let. Simptomi vključujejo slabšo šolsko uspešnost, duševne motnje, ataksijo, epileptične napade in demenco. Simptomi napredujejo, smrt nastopi 10–20 let po začetku bolezni.
- Oblika za odrasle. Simptomi odraslih se začne po 16. letu in lahko vključuje ataksijo, epileptične napade, nenormalno tresenje okončin (tresenje), oslabljeno koncentracijo, depresija, duševne motnje in demenca. Smrt običajno nastopi 6–14 let po pojavu simptomov.
MLD ni mogoče pozdraviti. Zdravljenje je simptomatsko in podporno. Presaditev kostnega mozga lahko v nekaterih primerih upočasni napredovanje bolezni. Pomemben napredek je bil dosežen z gensko terapijo na živalskih modelih z MLD in v kliničnih preskušanjih.
Wolmanova bolezenpoznan tudi kot pomanjkanje lizosomske kislinske lipazeje resna motnja shranjevanja lipidov, ki je običajno usodna pri starosti enega leta. Za to avtosomno recesivno motnjo je značilno kopičenje estrov holesterola (običajno transportna oblika holesterola) in trigliceridi (kemična oblika, v kateri so maščobe v telesu), ki se lahko znatno kopičijo in povzročijo poškodbe celic in tkanine.
Za to motnjo trpijo tako moški kot ženske. Dojenčki so ob rojstvu normalni in aktivni, vendar se hitro razvijejo progresivne duševne motnje, povečana jetra in močno povečana vranica, napihnjenost, težave s prebavili. zlatenica, anemija, bruhanje in usedline kalcija v nadledvične žlezezaradi česar se strdijo.
Druga vrsta pomanjkanje lizosomske kislinske lipaze je bolezen shranjevanja holesterolnih estrov. Ta izjemno redka bolezen se pojavi kot posledica shranjevanja estrov holesterola in trigliceridov v krvnih celicah, limfo in limfoidno tkivo. Pri otrocih v odrasli dobi se jetra povečajo, kar vodi v cirozo in kronična odpoved jeter. Otroci imajo lahko tudi usedline kalcija v nadledvičnih žlezah, v poznejših fazah bolezni pa se lahko razvije zlatenica.
Trenutno se vprašanje zamenjave encimov aktivno preučuje tako pri Wolmanovi bolezni kot pri kopičenju estra holesterola.
Pozor! Informacije so zgolj informativne narave in niso namenjene diagnosticiranju in predpisovanju zdravljenja. Vedno se posvetujte s svojim zdravnikom specialistom!


