Robinovův syndrom: co to je, příčiny, příznaky, léčba, prognóza
Obsah
- Co je Robinoffův syndrom?
- Klinické příznaky Robinovova syndromu
- Příčiny Robinovova syndromu
- Postižené populace
- Diagnostika
- Léčba Robinovova syndromu
- Předpověď
- Podobné poruchy
Co je Robinoffův syndrom?
Robinoffův syndrom je extrémně vzácná dědičná porucha, která ovlivňuje vývoj kostí a dalších částí těla. Existují dvě formy Robinovova syndromu, které se liší znaky a symptomy, závažností, typem dědičnosti a s nimi spojenými geny.
PrvníAutozomálně recesivní Robinovův syndrom je závažnější a je charakterizován:
- zkrácení dlouhých kostí v pažích a nohou;
- krátké prsty na rukou a nohou;
- kosti páteře ve tvaru páteře, což vede k abnormálnímu zakřivení páteře (kyfoskolióza);
- vypouštěcí nebo chybějící žebra;
- malý vzrůst;
- výrazné rysy obličeje.
Mezi další funkce může patřit:
- nedostatečný rozvoj genitálií;
- zubní problémy;
- vady ledvin nebo srdce;
- vývojové zpoždění.
Děti s druhá formaAutozomálně dominantní Robinovův syndrom má podobné příznaky, ale projevuje se mírněji. Anomálie páteře a žeber obvykle chybí a malý vzrůst je méně výrazný.
Někteří lidé s autozomálně dominantním Robinovovým syndromem mohou mít také zvýšenou kostní minerální denzitu (osteoskleróza).
Klinické příznaky Robinovova syndromu

Robinovův autosomálně recesivní syndrom je charakterizován:
- malý vzrůst;
- charakteristické rysy obličeje;
- kosterní abnormality a / nebo genitální abnormality.
Rozsah a závažnost klinických příznaků se liší od člověka k člověku.
Většina dětí s Robinoffovým syndromem má zkušenosti zpomalení růstu po poroducož má za následek malý až středně malý vzrůst. Většina dětí má normální inteligenci, ale asi 20 procent postižených může mít:
- mentální postižení;
- zpoždění při dosahování milníků;
- zpoždění ve vývoji jazykových dovedností.
Funkce obličeje se podobají známky vyvíjejícího se ploduV lékařské literatuře se tomu často říká „fetální obličej“. Charakteristické abnormality hlavy a oblasti obličeje mohou zahrnovat velká hlava (makrocefalie) s vypuklým čelem a nedostatečný rozvoj středního povrchu (hypoplazie středního povrchu).
Postižené děti mohou mít také:
- Široce rozmístěné oči (oční hypertelorismus), které vyčnívají z oběžných drah (vyboulené)
- neobvykle široká šikmá víčka (oční štěrbiny);
- malý obrácený nos s nozdrami se rozšířil dopředu;
- potopený příďový most;
- Nesprávně umístěné (tj. Nízko nasazené, obrácené dozadu) uši.
Některé postižené děti mohou mít:
- široká, trojúhelníková ústa, obrácená dolů, s dlouhou štěrbinou uprostřed horního rtu;
- malá brada;
- malá čelist (mikrognatie);
- gingivální hyperplazie.
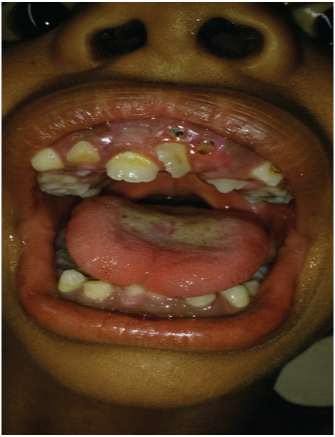
Mohou být také přítomny zubní abnormality, včetně:
- nesouosost zubů;
- zadní shlukování zubů;
- opožděná erupce sekundárních (trvalých) zubů.
Struktura měkkých tkání v zadní části krku (uvula) může být nedostatečně vyvinutá nebo abnormálně rozdělena (rozdvojená). Postižené děti mohou mít také rozštěp patra, abnormální svislou drážku nebo díru v horním rtu (rozštěp rtu) a / nebo omezený pohyb jazyka (ankyloglosie).
Ankyloglossia může přispět ke zpoždění ve vývoji jazykových dovedností. U většiny dětí jsou obličejové abnormality spojené s Robinovovým syndromem s věkem méně výrazné.
Abnormality kostry mohou zahrnovat:
- patologie kostí předloktí (poloměr a ulna), které vypadají abnormálně krátké a nedostatečně vyvinuté (brachymelia předloktí);
- neobvyklá odchylka strany palce předloktí (poloměr) v důsledku zkrácení poloměru (madelung zápěstí);
- neobvykle krátké prsty (brachydaktylie);
- trvalá fixace pátého prstu ve flexi (klinodaktylie);
- neobvykle malé ruce se širokými palci.
Koncové kosti (koncové falangy) velkých prstů mohou být abnormálně odděleny a / nebo kosti (falangy) prstů a prstů mohou být nedostatečně vyvinuté (hypoplastické). Další abnormality mohou zahrnovat:
- posun kyčelního kloubu;
- omezené prodloužení lokte, abnormální fúze nebo absence určitých žeber;
- abnormální zakřivení páteře ze strany na stranu (skolióza);
- nedostatečné rozvinutí jedné strany kostí (obratlů) ve střední (hrudní) části páteře (hemivertebra);
- fúze některých obratlů.
Přečtěte si také:Ehlers-Danlosův syndrom
Postižené děti mohou zažít abnormální prohloubení kostítvořící střed hrudníku („nálevkovitý hrudník“). Děti mohou mít také deformované (dysplastické) nehty a / nebo abnormality ve vzorcích hřebene kůže na prstech a dlaních.
Většina dětí s Robinoffovým syndromem také má genitální anomáliea některé mohou mít vnější genitálie, které nejsou jasně mužské nebo ženské (nejednoznačné genitálie). Pohlaví lze obvykle správně identifikovat v raném dětství. Mezi ženami Klitoris a vnější prodloužené záhyby kůže na obou stranách vaginálního otvoru (labia majora) mohou být nedostatečně vyvinuté (hypoplastické). U mužů penis může být abnormálně malý (mikropenis) a může být skryt pod okolní kůží; Navíc jedno nebo obě varlata nesmí sestoupit do šourku (kryptorchismus).
Zřídka mohou mít postižení muži abnormálně nízké hladiny testikulárních funkcí (částečné primární hypogonadismus, ale existuje normální vývoj sekundárních sexuálních charakteristik (například hrubý hlas, charakteristické vzorce růst vlasů, náhlé zvýšení růstu a vývoj varlat a šourku atd.), s výjimkou vytrvalosti mikropenis.
Postižené ženy vykazují normální funkci vaječníků (gonadální funkce) a normální plodnost.
Lidé s Robinoffovým syndromem mohou mít další tělesné postižení, jako například:
- zdvojnásobení ledvin;
- neobvyklé hromadění moči v ledvinách (hydronefróza);
- výčnělek částí tlustého střeva abnormálním otvorem ve výstelce břicha (tříselná kýla);
- vyboulení částí tlustého střeva přes břišní stěnu poblíž pupku (pupeční kýla).
Navíc přibližně 13 procent dětí s Robinoffovým syndromem má při narození srdeční vady (vrozené srdeční vady). V těchto případech je nejčastější srdeční vadou „obstrukce pravé komory“, při které je zablokován průtok krve z dolní srdeční komory (komory) v důsledku abnormálního zúžení (stenóza) nebo uzavření (atrézie) cévy vyplývající z komory (plicní kmen) a dělení na levou a pravou plicní tepny.
Příznaky spojené s touto srdeční vadou se velmi liší v závislosti na velikosti a umístění překážky. Některé děti s Robinoffovým syndromem mohou mít jiné srdeční problémy, včetně komplexních vrozených srdečních vad, které mohou vést k život ohrožujícím komplikacím.
Vzácně mohou být kojenci a děti s Robinoffovým syndromem náchylní k recidivujícím plicním infekcím (zápal plic). Ve vzácných, závažných případech bez vhodné léčby zápal plic může vést k život ohrožujícím komplikacím.
Dominantní a recesivní formy Robinovova syndromu mají mnoho stejných symptomů a fyzických příznaky (např. kraniofaciální abnormality, nízký vzrůst, malformace skeletu a hypoplazie genitálu orgány). Příznaky a fyzické příznaky spojené s recesivní formou však bývají závažnější.
Kojenci s recesivním Robinovovým syndromem vykazují četnější abnormality žeber (např. Abnormální posunutí, fúze a / nebo absence určitých žeber) a defekty postihující kosti páteře (obratle) než u dominantních kojenců nemoci. Kromě toho je závažnější krátký vzrůst, nedostatečný vývoj kostí předloktí (radiační hypoplazie) a anomálie prstů. Postižené děti mohou mít dislokaci hlavy jedné z kostí předloktí a u lidí s dominantní formou Robinovova syndromu je tato anomálie pozorována jen zřídka. Lidé s recesivní formou mohou mít také více trojúhelníkový tvar úst.
Variantní forma autozomálně dominantního Robinovova syndromu, osteosklerotická forma, se vyznačuje:
- zvýšená minerální hustota kostí, zejména v lebce;
- normální růst;
- velká hlava;
- a ztrátu sluchu, kromě typických známek a příznaků.
Přečtěte si také:Aceton v moči dítěte (acetonurie u dětí): co dělat, příčiny, příznaky, léčba
Příčiny Robinovova syndromu

Autozomálně recesivní a autozomálně dominantní formy Robinovova syndromu jsou způsobeny změnami (mutacemi) v různých genech.
Autozomálně recesivní typ k onemocnění dochází v důsledku mutací v genu ROR2což vede k nedostatku bílkovin ROR2. Bez tohoto proteinu je narušen vývoj dítěte, zejména tvorba kostry, srdce a genitálií.
Autozomálně dominantní typ k onemocnění dochází v důsledku mutací v genu WNT5A nebo gen DVL1, což vede k nedostatku specifických proteinů nezbytných pro normální vývoj. Osteosklerotická forma je způsobena mutace gen DVL1.
Malé procento dětí s Robinoffovým syndromem nemá mutace v žádném z těchto genů a příčina onemocnění zůstává neznámá.
Většina genetických chorob je dána stavem dvou kopií genu, jedné od otce a druhé od matky. Recesivní genetické poruchy nastávají, když člověk zdědí dvě kopie abnormálního genu pro stejný znak, jednu od každého rodiče. Pokud osoba zdědí jeden normální gen a jeden gen pro tuto nemoc, bude osoba přenášet nemoc, ale obvykle nevykazuje žádné příznaky. Riziko pro dva rodiče nosiče, kteří oba předají pozměněný gen a nakazí dítě, je 25% s každým těhotenstvím. Riziko narození dítěte, které bude nositelem, jako rodič, je při každém těhotenství 50%. Dítě má 25% šanci na získání normálních genů od obou rodičů. Riziko je stejné pro muže i ženy.
Rodiče, kteří jsou blízkými příbuznými (incest), mají vyšší šanci mít nemocné dítě než rodiče, kteří nejsou ve spojení.
Dominantní genetické poruchy se vyskytují, když je k vyvolání konkrétní choroby zapotřebí pouze jedna kopie abnormálního genu. Abnormální gen může být zděděn po kterémkoli z rodičů, nebo může být důsledkem nové mutace (změny genu) u postižené osoby. Riziko přenosu abnormálního genu z postiženého rodiče na potomstvo je 50% pro každé těhotenství. Riziko je stejné pro muže i ženy.
U některých lidí je tato porucha způsobena spontánní genetickou mutací, která se vyskytuje ve vejci nebo spermatu. V takových situacích není porucha zděděna po rodičích.
Postižené populace
Autosomálně recesivní syndrom byl hlášen u méně než 200 lidí v rodinách z různých etnických skupin.
Autosomálně dominantní syndrom byl hlášen v méně než 50 rodinách.
Diagnostika
Diagnóza Robinoffova syndromu se obvykle provádí krátce po narození na základě fyzických nálezů, včetně nízké postavy, anomálií končetin a genitálií a obličejových rysů.
K potvrzení diagnózy autozomálně recesivního Robinovova syndromu je k dispozici molekulárně genetické testování genu ROR2. Molekulárně genetické testování mutací v genu WNT5A a gen DVL1 k dispozici k potvrzení diagnózy autozomálně dominantního typu onemocnění.
Léčba Robinovova syndromu
Léčba Robinovova syndromu se zaměřuje na specifické příznaky, které každý člověk zažívá. Léčba může vyžadovat koordinované úsilí týmu specialistů. Pediatři, specialisté na léčbu poruch skeletu (ortopedové), chirurgové, specialisté na diagnostiku a léčbu srdečních problémů (kardiologové), fyzioterapeuti a / nebo jiní poskytovatelé zdravotní péče mohou potřebovat systematické a komplexní plánování léčby zraněné dítě.
U postižených dětí s nejednoznačnými genitáliemi lze pohlaví správně určit klinickým vyšetřením během novorozeneckého období. U některých dětí může být provedena operace a / nebo mohou být přijata jiná opatření k nápravě kryptorchismu a / nebo jiných genitálních abnormalit. Kromě toho se někdy podávají k léčbě mikropenis hormonální doplňky, jako je lidský choriový gonadotropin a / nebo testosteron, a to zlepšením délky penisu a objemu varlat. U některých pacientů bylo zjištěno, že nedostatek růstového hormonu pozitivně reaguje na léčbu růstovým hormonem. Tyto formy hormonální terapie však musí být pečlivě sledovány dětským endokrinologem.
Speciální cvičení a / nebo operace mohou být užitečné při léčbě některých poruch páteře. Může být také provedena operace k nápravě tříselné kýly, určitých kraniofaciálních abnormalit, závažného zakřivení páteře (skoliózy) sekundárně k ve vztahu k abnormalitám hemivertebry a žeber, zcela nebo částečně spojené prsty / prsty (syndaktylie), rozštěp rtu / patra a / nebo jiné defekty rozvoj. Rovnátka, zubní chirurgie a / nebo jiné podpůrné terapie mohou pomoci opravit nevyrovnané zuby a / nebo jiné orální abnormality.
Přečtěte si také:Dyslexie
Děti a děti s Robinoffovým syndromem musí podstoupit důkladnou lékařskou prohlídku, aby byla zajištěna včasnost identifikace a okamžitá vhodná léčba srdečních patologií, které by s tím mohly být potenciálně spojeny porucha. Postižení kojenci a děti by také měli být pečlivě sledováni, aby se předešlo a / nebo okamžitě detekovala plicní infekce (pneumonie) a poskytla se rychlá a vhodná léčba.
Včasná intervence je nezbytná pro to, aby děti s Robinovovým syndromem dosáhly svého potenciálu. Speciální služby, které mohou být užitečné pro postižené děti, mohou zahrnovat nápravu vzdělávání, sociální podpora, pohybová terapie a / nebo jiné lékařské, sociální a / nebo profesionální služby.
Předpověď
Prognóza Robinovova syndromu je obecně dobrá, ale závažnost srdeční poruchy může ovlivnit délku života.
Podobné poruchy
Příznaky následujících poruch mohou být podobné jako u Robinovova syndromu. Srovnání jsou užitečné pro diferenciální diagnostiku:
AchondroplasieJe dědičná porucha charakterizovaná abnormálně krátkými pažemi a nohami a nízkým vzrůstem (zakrslost krátkých končetin), abnormálními rysy obličeje a / nebo malformacemi kostry. Obličejové rysy mohou zahrnovat abnormálně velkou hlavu (makrocefalii), neobvyklé vyboulení čela, nízký nosní můstek a / nebo nedostatečné rozvinutí středního povrchu (hypoplazie středního obličeje).
Kosterní malformace mohou zahrnovat neobvykle krátké prsty na rukou a nohou (brachydaktylie), sagitální ohyb páteře, který je vyklenutý dopředu (lordóza), zúžení (stenóza) páteře. Mezi další poruchy mohou patřit problémy s prodloužením lokte a kyčle, snížený svalový tonus (hypotenze) a / nebo časté infekce středního ucha (zánět středního ucha).
Achondroplasie je autozomálně dominantní genetická porucha.
Aarskogův syndrom je velmi vzácná dědičná porucha charakterizovaná anomáliemi hlavy a obličeje (kraniofaciální) oblast, malý až středně malý vzrůst, kosterní malformace a / nebo genitální anomálie. Obličejové rysy mohou zahrnovat široce rozmístěné oči (oční hypertelorismus), nakloněné dolů záhyby očních víček (oční štěrbiny), malý nos s nosními dírkami a / nebo nedostatečné rozvinutí horní čelisti (hypoplazie horní čelist).
Kosterní malformace mohou zahrnovat abnormálně krátké prsty (brachydaktylie), trvalou fixaci pátých prstů ve flexi (klinodaktylie) a / nebo neobvykle široké palce. Genitální abnormality mohou zahrnovat abnormální kožní záhyb rozkládající se kolem základny penisu a / nebo neschopnost jednoho nebo obou varlat sestoupit do šourku (kryptorchismus). Některé postižené děti mají lehkou mentální retardaci.
Aarskogův syndrom je genetická porucha spojená s X.
Omodysplázie Je vzácná dědičná kostní porucha, kterou lze charakterizovat abnormální krátkostí některých dlouhých kostí paží (např. Pažní kosti) a nohou (tj. Stehna) kosti), nízkého vzrůstu a charakteristických rysů obličeje, včetně sníženého nosního můstku, široké základny nosu a / nebo neobvykle dlouhé svislé drážky (rýhy) ve středu horní části rty.
Kromě toho mohou mít postižené děti abnormální oddělení dvou kostí předloktí (radiální diastáza), dislokace radiální hlavy a / nebo nedostatečný vývoj (hypoplazie) zaoblené kostní hmoty (kondylu) na konci rameno.
Omodysplázii lze zdědit jako autosomálně recesivní nebo autozomálně dominantní genetický stav.