
Lipoidóza (lysozomální onemocnění ukládání lipidů): co to je, příklady
Pozornost! Informace slouží pouze pro informační účely a nejsou určeny k diagnostice a předepisování léčby. Vždy se poraďte se svým odborným lékařem!
Obsah
- Co jsou lysozomální střádavá onemocnění?
- Co jsou to lipidy?
- Jak se lipoidóza dědí?
- Typy a příznaky lysozomálních střádavých chorob
Co jsou lysozomální střádavá onemocnění?
Lysozomální střádavá onemocnění (lipoidóza), jsou skupinou dědičných metabolických onemocnění, při kterých se škodlivé množství tukových látek (lipidů) hromadí v různých buňkách a tkáních těla.
Lidé s těmito poruchami buď nevytvářejí dostatek jednoho z enzymů potřebných k rozkladu (metabolizaci) lipidů, nebo produkují enzymy, které nefungují správně.
Toto nadměrné hromadění tuku může časem vést k trvalému poškození buněk a tkání, zejména v mozku, periferním nervovém systému (nervy od míchy po zbytek tělo), játra, slezina a kostní dřeně.
Co jsou to lipidy?
Lipidy jsou látky podobné tuku, které jsou důležitými součástmi membrán uvnitř buněk a mezi nimi a v myelinové pochvě, která pokrývá a chrání nervy. Lipidy zahrnují oleje, mastné kyseliny, vosky, steroidy (jako je cholesterol a estrogen) a další příbuzné sloučeniny.
Tyto tukové látky se přirozeně ukládají v buňkách, orgánech a tkáních těla. Drobná tělíska v buňkách, nazývaná lysozomy, pravidelně přeměňují nebo metabolizují lipidy a proteiny na menší složky, které tělu poskytují energii. Poruchy, při kterých intracelulární materiál, který nemůže být metabolizován, zůstává v lysozomech, se nazývají lysozomální střádavá onemocnění.
Kromě onemocnění spojených s akumulací lipidů zahrnují další lysozomální onemocnění spojená s akumulací mukolipidózy, při kterých se hromadí buňky a tkáně. nadměrné množství lipidů s navázanými molekulami cukru a také mukopolysacharidózy, ve kterých se hromadí nadměrné množství velkého komplexního cukru molekul.
Jak se dědí lipoidóza?
Lysozomální střádavá onemocnění se dědí od jednoho nebo obou rodičů, kteří jsou nositeli defektního genu, který reguluje specifický enzym metabolizující lipidy ve třídě buněk v těle. Mohou být zděděny dvěma způsoby:
- Autozomálně recesivní dědičnost nastává, když oba rodiče nesou a předávají kopii defektního genu, ale žádný z rodičů není touto poruchou postižen. Každé dítě narozené těmto rodičům má 25procentní pravděpodobnost, že zdědí obě kopie defektního genu, 50 procent pravděpodobnost, že bude nosičem podobným jeho rodičům, a 25procentní pravděpodobnost, že nezdědí žádnou kopii vadného gen. Děti obou pohlaví mohou být zděděny autozomálně recesivním způsobem.
- X-vázaná (nebo pohlaví-příbuzná) recesivní dědičnost nastává, když matka nese postižený gen na X chromozomu. Na určování pohlaví se podílejí chromozomy X a Y. Ženy mají dva chromozomy X, zatímco muži mají jeden chromozom X a jeden chromozom Y. Synové nositelek mají 50procentní šanci, že zdědí a ovlivní poruchu, protože synové obdrží jeden chromozom X od své matky a jeden chromozom Y od svého otce. Dcery mají 50procentní šanci, že zdědí postižený chromozom X od své matky a jsou přenašečkami nebo mírně postižené. Postižení muži nepřenášejí nemoc na své syny, ale jejich dcery budou přenašečkami a přenašečkami nemoci.
Typy a příznaky lysozomálních střádavých chorob
Gaucherova nemoc - nejčastější z lysozomálních onemocnění. Onemocnění je způsobeno nedostatkem enzymu glukocerebrosidázy, který vede k akumulaci glukocerebrosidu v mnoha tkáních. Tukový materiál se může hromadit v mozku, slezině, játrech, ledvinách, plicích a kostní dřeni.
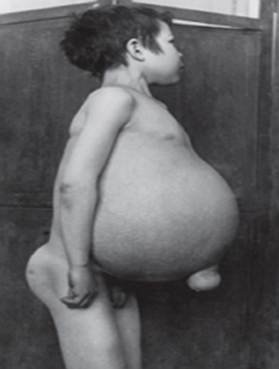
Příznaky mohou zahrnovat poškození mozku, zvětšení sleziny a jater, zhoršená funkce jater, kosterní poruchy a poškození kostí, které může způsobit bolest a zlomeniny, otoky lymfatických uzlin a (někdy) přilehlých kloubů, nadýmání, hnědý odstín pleti, anémie, nízký počet krevních destiček a žluté skvrny na očích.
Nejvážněji postižení jedinci mohou být také náchylnější k infekcím. Nemoc postihuje stejně muže i ženy.
Gaucherova choroba má tři obecné klinické podtypy:
- Typ 1 (Nebo ne neuronopatický typu) je nejběžnější formou onemocnění ve Spojených státech a v Evropě. Mozek není postižen, ale může dojít k poškození plic a ve vzácných případech i ledvin. Příznaky mohou začít brzy v životě nebo v dospělosti a zahrnují zvětšená játra a závažné zvětšení sleziny, což může vést k prasknutí a dalším komplikacím. Kosterní slabost a onemocnění kostí mohou být rozsáhlé. Lidé v této skupině se obvykle snadno tvoří modřiny kvůli nízkému počtu krevních destiček v krvi. Mohou také pociťovat únavu v důsledku anémie. V závislosti na počátku a závažnosti onemocnění mohou lidé s typ 1 se může dožít dospělosti. Mnoho postižených má mírné onemocnění nebo nemusí vykazovat žádné příznaky. Ačkoli 1typ Gaucherova choroba je běžná mezi lidmi aškenázského židovského původu a může postihnout lidi všech etnických původů.
- Typ 2 (nebo akutní dětský neuropatický Gaucherova choroba) obvykle začíná do 3 měsíců po narození. Příznaky zahrnují rozsáhlé a progresivní poškození mozku, spasticitu, záchvaty, ztuhlé končetiny, zvětšená játra (jaterní hepatomegalie) a slezina (splenomegalie), abnormální pohyby očí a špatná schopnost sát a polykat. Postižené děti obvykle umírají do 2 let věku.
- Typ 3 (chronický neuronopatický forma) může začít kdykoli v dětství nebo dokonce v dospělosti. Je charakterizována pomalu progredujícími, ale méně závažnými neurologickými příznaky ve srovnání s akutní Gaucherovou chorobou 2 druhy. Mezi hlavní příznaky patří poruchy pohybu očí, kognitivní deficity, špatná koordinace, záchvaty, zvětšení sleziny a/nebo jater, poruchy skeletu, poruchy krve včetně anémie a problémy s dýchání. Téměř každý s Gaucherovou chorobou 3 druhy, kdo dostává enzymovou substituční terapii, dosáhne dospělosti.
Pro pacienty 1. a 3. typ Enzymová substituční terapie, podávaná intravenózně každé dva týdny, může dramaticky zmenšit velikost jater a sleziny, snížit kosterní abnormality a zvrátit další projevy. Úspěšná transplantace kostní dřeně léčí neurologické projevy onemocnění. Tento postup však přináší značná rizika a u jedinců s Gaucherovou chorobou se provádí jen zřídka.
Ve vzácných případech může být vyžadován chirurgický zákrok k odstranění celé nebo části sleziny (pokud má osoba velmi nízký počet krevních destiček nebo když zvětšený orgán výrazně ovlivňuje pohodlí osoby). Krevní transfuze může být přínosem pro některé lidi s anémií. Jiní mohou potřebovat operaci kloubní náhrady ke zlepšení mobility a kvality života. V současné době neexistuje účinná léčba poškození mozku, které se může objevit u lidí s Gaucherovou chorobou typu 2 a 3.
Niemann-Pickova choroba je skupina autozomálně recesivních poruch způsobených hromaděním tuku a cholesterolu v buňkách jater, sleziny, kostní dřeně, plic a v některých případech i mozku.
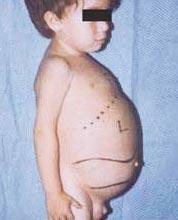
Neurologické komplikace mohou zahrnovat ataxii (nedostatek svalové koordinace, která může ovlivnit chůzi, psaní a jídlo, mimo jiné), paralýza oka, degenerace mozku, problémy s učením, spasticita, potíže s krmením a polykáním, nezřetelná řeč, ztráta svalového tonusu, přecitlivělost na dotek a určitá zákal rohovky v důsledku nadměrného hromadění materiálů. U 50 % postižených se kolem středu sítnice vytvoří charakteristické třešňově červené halo, které může vidět lékař speciálním nástrojem.
Niemann-Pickova choroba je klasifikována do tří obecných podtypů:
- Typ A, nejtěžší forma, začíná v raném dětství. Děti vypadají po narození normálně, ale do 6 měsíců se rozvine hluboké poškození mozku, zvětšená játra a slezina a zduřené lymfatické uzliny a uzliny pod kůží (xantomy). Slezina se může zvětšit na 10násobek své normální velikosti a může prasknout a způsobit krvácení. Tyto děti postupně slábnou, ztrácejí motorické funkce, mohou být anemické a jsou náchylné k opakovaným infekcím. Zřídka žijí déle než 18 měsíců. Tato forma onemocnění je nejčastější v židovských rodinách.
- Typ B(nebo juvenilní nástup) obvykle neovlivňuje mozek, ale většina dětí se vyvíjí ataxiepoškození nervů opouštějících míchu (periferní neuropatie) a plicní potíže, které progredují s věkem. Pro dospívající je typické zvětšení jater a sleziny. Lidé s typ B mohou žít relativně dlouho, ale mnoho z nich vyžaduje celoživotní oxygenoterapii kvůli poškození plic. Niemann-Pick typy A a B jsou výsledkem akumulace tukové látky zvané sfingomyelin v důsledku nedostatku enzymu zvaného sfingomyelináza.
- Typ C se může objevit v raném věku nebo se rozvinout během dospívání nebo dokonce v dospělosti. Niemann-Pickova chorobatyp C není způsobena deficiencí sphlingomyelinázy, ale nedostatkem proteinů NPC1 nebo NPC2. V důsledku toho se různé lipidy a zejména cholesterol hromadí v nervových buňkách a způsobují jejich špatnou funkci. Postižení mozku může být rozsáhlé, což má za následek neschopnost dívat se nahoru a dolů, potíže s chůzí a polykáním, progresivní ztrátu sluchu a progresivní demence. Lidé s typ C mají pouze mírné zvětšení sleziny a jater. Očekávaná délka života lidí s typ C se značně liší. Někteří lidé zemřou v dětství, zatímco jiní, kteří jsou méně postiženi, mohou žít i v dospělosti.
V současnosti neexistuje žádný lék na Niemann-Pickovu chorobu. Léčba je podpůrná. Děti obvykle umírají na infekci nebo progresivní neurologické ztráty. U několika pacientů se vyskytly případy transplantace kostní dřeně typ B se smíšenými výsledky.
Fabryho nemoctaké známý jako nedostatek alfa-galaktosidázy-Azpůsobuje hromadění tukové hmoty v autonomním nervovém systému (ta část nervového systému, která řídí mimovolní funkce, jako je dýchání a srdeční tep), v očích, ledvinách a kardiovaskulárním systému Systém.
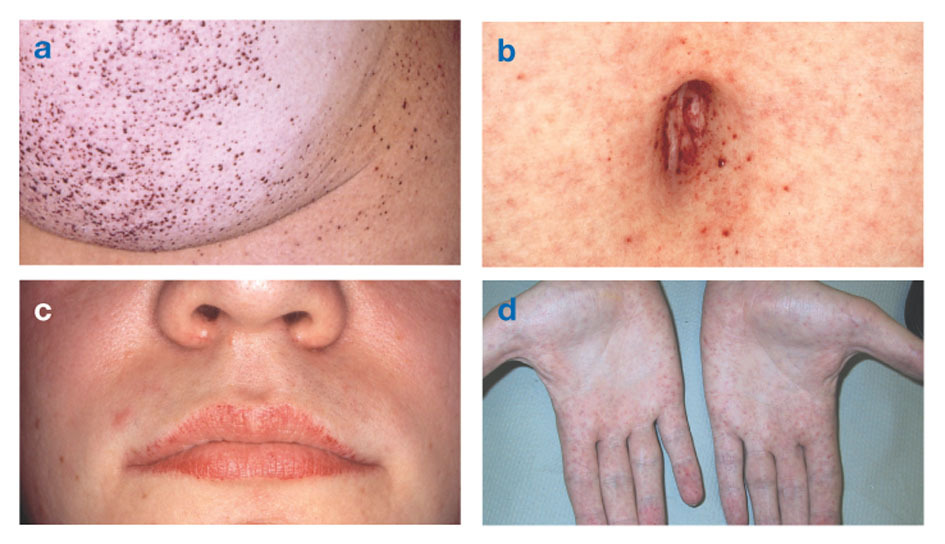
Fabryho choroba je jediné X-vázané lipidové onemocnění. Onemocnění postihuje především muže, i když mírnější a variabilnější forma se vyskytuje u žen. Vzácně mají postižené ženy závažné příznaky podobné těm, které se vyskytují u mužů s touto poruchou. K nástupu symptomů obvykle dochází v dětství nebo dospívání.
Neurologické příznaky zahrnují palčivou bolest v pažích a nohou, která se zhoršuje v horkém počasí nebo později cvičení, stejně jako hromadění přebytečného materiálu v průhledných vrstvách rohovky (což vede k zakalení, ale bez změny vidění). Hromadění tuku ve stěnách krevních cév může narušit oběh a vystavit člověka riziku mrtvice nebo infarkt. Mezi další příznaky patří zvětšené srdce, progresivní selhání ledvincož vede ke konečnému stádiu onemocnění ledvin, gastrointestinálním problémům, sníženému pocení a horečce.
Angiokeratomy (malé, nerakovinné, červenofialové vyvýšené skvrny na kůži) se mohou vyvinout v dolní části trupu a stávají se častějšími s věkem.
Lidé s Fabryho chorobou často umírají předčasně na komplikace způsobené srdeční selhání, selhání ledvin nebo mrtvice. Léky jako fenytoin a karbamazepin jsou často předepisovány k léčbě bolesti, která doprovází Fabryho chorobu, ale nikoli k jejímu vyléčení. Metoklopramid nebo Lipisorb (doplněk stravy) mohou zmírnit gastrointestinální potíže se často vyskytuje u lidí s Fabryho chorobou a někteří lidé mohou potřebovat transplantaci ledvin nebo dialýza. Náhrada enzymů může u některých lidí s Fabryho chorobou snížit ukládání, zmírnit bolest a zachovat funkci orgánů.
Farberova nemoc také známý jako Farberova lipogranulomatóza, popisuje skupinu vzácných autozomálně recesivních poruch, které způsobují hromadění tukové hmoty v kloubech, tkáních a centrálním nervovém systému. Postihuje muže i ženy. Onemocnění obvykle začíná v raném dětství, ale může se objevit i později v životě.
Rozvíjí se děti s klasickou Farberovou chorobou neurologické příznaky, které mohou zahrnovat zvýšenou ospalost a letargii a problémy s polykání. Postižena mohou být také játra, srdce a ledviny. Další příznaky mohou zahrnovat kontraktury kloubů (chronické zkrácení svalů nebo šlach kolem kloubů), zvracení, artritidu, zvětšení lymfatické uzliny, záněty kloubů, chrapot a hrudky pod kůží, které se při postupu ztlušťují kolem kloubů nemocí.
Postižení lidé s dýchacími potížemi mohou potřebovat zavedení dýchací trubice. Většina dětí s tímto onemocněním zemře ve věku 2 let, obvykle od plicní onemocnění. U jedné z nejtěžších forem onemocnění lze krátce po narození diagnostikovat zvětšená játra a slezinu. Děti narozené s touto formou onemocnění obvykle umírají do 6 měsíců.
Farberova choroba je způsobena nedostatkem enzymu zvaného ceramidáza. V současné době neexistuje žádná specifická léčba Farberovy choroby. K úlevě od bolesti mohou být předepsány kortikosteroidy. Transplantace kostní dřeně může redukovat granulomy (malé masy zanícené tkáně) u lidí s mírným zánětem nebo u pacientů bez plicních či nervových komplikací. U starších lidí lze granulomy zmenšit nebo chirurgicky odstranit.
Gangliosidóza sestává ze dvou různých skupin genetických onemocnění. Oba jsou autozomálně recesivní a postihují stejně muže i ženy.
GM1 gangliosidóza.
Gangliosidóza GM1 je způsobena nedostatkem enzymu beta-galaktosidázy, což má za následek abnormální akumulaci kyselé lipidové materiály, zejména v nervových buňkách v centrálním a periferním nervu systémy. Gangliosidóza GM1 má tři klinické projevy:
- GM1 (nejzávažnější podtyp, vyskytující se brzy po narození) může zahrnovat neurodegeneraci, křeče, zvětšená játra a slezinu, hrubost obličejových rysů, kosterní nepravidelnosti, ztuhlost kloubů, nadýmání, svalová slabost, přehnaná úleková reakce a problémy s chůze. Asi u poloviny postižených se v oku objeví třešňově červené skvrny. Děti mohou být hluché a slepé ve věku 1 a často umírají ve věku 3 let na srdeční komplikace nebo zápal plic.
- Pozdní dítě Gangliosidóza GM1 obvykle začíná ve věku od 1 do 3 let. Neurologické příznaky zahrnují ataxii, záchvaty, demenci a potíže s mluvením.
- GM1 gangliosidóza vyvíjí se ve věku 3 až 30 let. Příznaky zahrnují snížení svalové hmoty (svalová atrofie), neurologické komplikace, které jsou méně závažné a postupují pomaleji než jiné formy poruchy, zákal rohovky u některých lidí a trvalé svalové kontrakce, které způsobují kroucení a opakované pohyby nebo abnormální držení těla (dystonie). Angiokeratomy se mohou vyvinout v dolní části těla. Velikost jater a sleziny je u většiny postižených lidí normální.
GM2 gangliosidóza.
Gangliosidóza GM2 také způsobuje hromadění přebytečných kyselých tukových látek v tkáních a buňkách, především nervových. Tyto poruchy jsou důsledkem nedostatku enzymu beta-hexosaminidázy. Poruchy GM2 zahrnují:
- Tay-Sachsova nemoc (také známá jako GM2 gangliosidóza varianta B) a její varianty jsou způsobeny deficitem enzymu hexosaminidázy A. Postižené děti se během prvních měsíců života vyvíjejí normálně. Příznaky začínají ve věku 6 měsíců a zahrnují progresivní ztrátu duševní kapacity, demenci, snížený oční kontakt, zvýšený úleková reakce na hluk, progresivní ztráta sluchu vedoucí k hluchotě, potíže s polykáním, slepota, třešňové červené skvrny na sítnici a některé ochrnutí. Záchvaty mohou začít ve druhém roce života dítěte. Děti se mohou nakonec stát závislými na přívodní trubici a často umírají ve věku 4 let na opakující se infekce. Bohužel neexistuje žádná speciální léčba. Antikonvulziva mohou zpočátku kontrolovat záchvaty. Mezi další podpůrné léčby patří správná výživa a hydratace a metody pro udržení otevřených dýchacích cest. Vzácná forma poruchy tzv Tay-Sachsova choroba s pozdním nástupemse vyskytuje u lidí ve věku 20 až 30 let a je charakterizována nestabilní chůzí a progresivním neurologickým zhoršováním.
- Sandhoffova choroba (Volba AB) je závažná forma Tay-Sachsovy choroby. Nástup obvykle nastává ve věku 6 měsíců a není omezen na žádnou etnickou skupinu. Neurologické příznaky mohou zahrnovat progresivní zhoršování centrálního nervového systému, motorické slabost, časná slepota, těžká vyděšená reakce na zvuk, spasticita, šok nebo záškuby svaly (myoklonus), epilepsie, abnormálně zvětšená hlava (makrocefalie) a červené skvrny v oku. Další příznaky mohou zahrnovat časté infekce dýchacích cest, srdeční šelest, panenkové rysy obličeje a zvětšení jater a sleziny. Neexistuje žádná specifická léčba Sandhoffovy choroby. Stejně jako u Tay-Sachsovy choroby zahrnuje podpůrná péče neustálé udržování dýchacích cest otevřených a také správnou výživu a hydrataci. Antikonvulziva mohou zpočátku kontrolovat epilepsii (záchvaty).
Krabbeho nemoc(také známý jako globulární buněčná leukodystrofie a galaktosylceramidová lipidóza) je autozomálně recesivní onemocnění způsobené nedostatkem enzymu galaktocerebrosidázy. Onemocnění nejčastěji postihuje kojence s nástupem před 6. měsícem věku, ale může se objevit během dospívání nebo dospělosti.
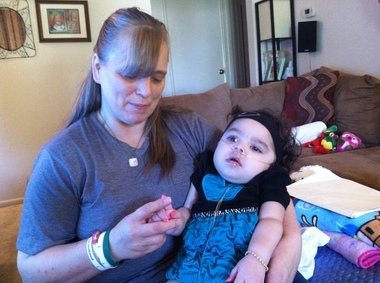
Hromadění nestrávených tuků ovlivňuje růst ochranného obalu nervu (myelinová pochva) a způsobuje vážné poškození mentálních a motorických schopností. Mezi další příznaky patří svalová slabost, snížená schopnost svalů natahovat se (hypotonie), svalové napětí (spasticita), náhlý šok nebo záškuby končetin (myoklonické záchvaty), podrážděnost, nevysvětlitelná horečka, hluchota, slepota, paralýza a potíže s polykáním. Může dojít i k dlouhodobému hubnutí.
Onemocnění lze diagnostikovat testováním enzymů a identifikací charakteristických skupin buněk do globoidních těles v bílé hmotě mozku, demyelinizace nervů a degenerace a destrukce mozkových buněk. U kojenců je onemocnění obvykle smrtelné před dosažením 2 let věku. Lidé s pokročilejší formou onemocnění mají mírnější průběh onemocnění a žijí výrazně déle.
Nebyla vyvinuta žádná specifická léčba Krabbeho choroby, i když časná transplantace kostní dřeně může některým lidem pomoci. Lidé s pokročilejší formou onemocnění mají mírnější průběh onemocnění a žijí výrazně déle.
Metachromatická leukodystrofie, nebo MLD, je skupina poruch vyznačujících se hromaděním tukových látek v bílé hmotě centrálního nervového systému a v periferních nervech a do určité míry v ledvinách. Podobně jako Krabbeho choroba ovlivňuje MLD myelin, který pokrývá a chrání nervy. Tato autozomálně recesivní porucha je způsobena deficitem enzymu arylsulfatázy A. Tato porucha postihuje muže i ženy.
Metachromatická leukodystrofie má tři charakteristické formy: pozdní kojenec, juvenilní a dospělý.
- Pozdní kojenecká MLD obvykle začíná mezi 12 a 20 měsíci po narození. Děti mohou zpočátku vypadat normálně, ale mají potíže s chůzí a sklonem k pádu, doprovázeným občasnou bolestí paží a nohou. progresivní ztráta zraku vedoucí ke slepotě, opoždění vývoje a ztrátě dříve získaných milníků, poruchy polykání, záchvaty a demence do 2 let. U dětí se také vyvíjí progresivní ochabování svalů a slabost a nakonec ztrácejí schopnost chodit. Většina dětí s touto formou poruchy umírá do věku 5 let.
- Juvenilní MLD obvykle začíná ve věku od 3 do 10 let. Příznaky zahrnují zhoršený školní prospěch, mentální postižení, ataxii, záchvaty a demenci. Příznaky progredují, smrt nastává 10–20 let po nástupu onemocnění.
- Dospělá forma. Příznaky Dospělí nástup po 16. roce věku a může zahrnovat ataxii, záchvaty, abnormální třes končetin (třes), poruchu koncentrace, Deprese, duševní poruchy a demence. Smrt obvykle nastává 6–14 let po nástupu příznaků.
MLD nelze vyléčit. Léčba je symptomatická a podpůrná. Transplantace kostní dřeně může v některých případech oddálit progresi onemocnění. Významného pokroku bylo dosaženo s genovou terapií na zvířecích modelech s MLD a v klinických studiích.
Wolmanova nemoctaké známý jako nedostatek lysozomální kyselé lipázyje závažná porucha ukládání lipidů, která je obvykle smrtelná ve věku 1 roku. Tato autozomálně recesivní porucha je charakterizována hromaděním esterů cholesterolu (obvykle transportní forma cholesterolu) a triglyceridy (chemická forma, ve které existují tuky v těle), které se mohou významně hromadit a způsobit poškození buněk a tkaniny.
Touto poruchou trpí muži i ženy. Děti jsou při narození normální a aktivní, ale rychle se u nich rozvine progresivní mentální poškození, zvětšená játra a značně zvětšená slezina, nadýmání, gastrointestinální problémy. žloutenka, anémie, zvracení a usazování vápníku v nadledvinkyzpůsobí jejich ztvrdnutí.
Jiný typ Nedostatek lysozomální kyselé lipázy je onemocnění z ukládání esterů cholesterolu. Toto extrémně vzácné onemocnění vzniká v důsledku ukládání esterů cholesterolu a triglyceridy v krvinkáchlymfatické a lymfoidní tkáně. U dětí v dospělosti se játra zvětší, což vede k cirhóze a chronický selhání jater. Děti mohou mít také usazeniny vápníku v nadledvinách a v pozdějších fázích onemocnění se může rozvinout žloutenka.
V současné době je problematika enzymové substituce aktivně studována jak u Wolmanovy choroby, tak u akumulace esteru cholesterolu.
Pozornost! Informace slouží pouze pro informační účely a nejsou určeny k diagnostice a předepisování léčby. Vždy se poraďte se svým odborným lékařem!


