Primäre pulmonale Hypertonie
 primäre pulmonale Hypertonie( PPH) oder Syndrom Aerza-Arilago - die Krankheit ist selten und kommt in 1-2 Fällen von einer Million. Die Diagnose der Krankheit ist schwierig, da die Symptome ähnlich wie bei den häufigeren Krankheiten sind. Die Ätiologie ist unbekannt, aber nach jüngsten Daten bezieht PLG erbliche Krankheiten, die mit einer genetischen Erwartung( in BMPR2 Gen markiert).Das heißt, mit jeder Generation gibt es eine Manifestation der Krankheit in einem früheren Alter. Meist wirkt sich die Krankheit Frauen jeden Alters. Es gibt eine familiäre und sporadische Formen der Krankheit.
primäre pulmonale Hypertonie( PPH) oder Syndrom Aerza-Arilago - die Krankheit ist selten und kommt in 1-2 Fällen von einer Million. Die Diagnose der Krankheit ist schwierig, da die Symptome ähnlich wie bei den häufigeren Krankheiten sind. Die Ätiologie ist unbekannt, aber nach jüngsten Daten bezieht PLG erbliche Krankheiten, die mit einer genetischen Erwartung( in BMPR2 Gen markiert).Das heißt, mit jeder Generation gibt es eine Manifestation der Krankheit in einem früheren Alter. Meist wirkt sich die Krankheit Frauen jeden Alters. Es gibt eine familiäre und sporadische Formen der Krankheit.
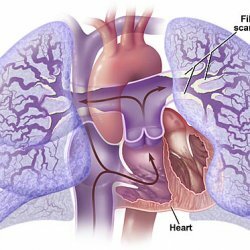
PPH wird durch erhöhten Druck im Lungenkreislauf, begleitet von Veränderungen in präkapillären Pulmonalarterien und thrombotischen Läsionen charakterisiert. In Abwesenheit von chronischen Herzerkrankungen und chronischer thromboembolischer Krankheit schreitet die Krankheit fort, in relativ kurzen Zeit entstandene( 2-8 Jahre) zu Rechtsherzversagen und Tod.
Krankheit verläuft drei
- Schritt 1 Schritt. Klinisch praktisch manifestiert, gibt es nur ein wenig kurzatmig nach dem Training und ist oft aufgrund mangelnder Ausbildung;2
- Schritt. Gekennzeichnet durch eine Abnahme der Herzleistung. Manifestierte sich in Form von Hypoxämie, Dyspnoe, erscheint Synkope. Gehalten stabil hohen Druck in der Lungenarterie;3
- Schritt. Es wird durch Rechtsherzversagen, der Rückgang der Herzleistung, venöse Stauung und periphere Ödeme charakterisiert. Ausführungsform
Manifestationen
- thrombotische Erkrankungen, zu microthromboembolism zu bilden;Plexogenic
- mit Bündel tunica muscularis aneurismal Dilatation von Arteriolen und Arteriolen;
- venoklyuzivny mit venöser Stauung in den Lungen.
pathologischen Veränderungen bei der Autopsie können pathologische Veränderungen des rechten Herzens und der Lunge erkennen. Die großen Gefäße sind sklerotische Plaques. Da die Prozessschicht kleine Lungenarterien, hypertrophe Muskels beinhaltet, gibt Fibrose und Intimahyperplasie. Manchmal fand es nekrotisierende Lungenarterie. Die Anzahl der Arteriolen und Kapillaren verringert wird, durch Basalmembran Verdickungs- und Hypertrophie der alveolaren Kapillaren medialen Pulmonalarterie charakterisiert.
PLG hat eine gewisse Ähnlichkeit mit der wiederkehrenden Lungenembolie. Der Unterschied besteht darin, dass, wenn rekanalisierten Thromboembolien Thromboembolien erkannt, aber keine plexiform Änderungen. Außerdem gibt es Ähnlichkeit mit venookklyuzionnoy Lungenerkrankung.
Prädisposition
Wie oben erwähnt, PLG - eine Erbkrankheit.prädisponieren auch zu seiner Entwicklung, wie der Pathologie:
- Kollagenosen,
- HIV,
- Zirrhose, Bluthochdruck begleitet poratalnoy,
- rheumatoide Arthritis,
- unidentifizierten Thromboembolie,
- pulmonale vaskuläre Embolisation.
Wenn wir über Risikofaktoren sprechen, nach jüngsten Daten, lösen die Entwicklung dieser Krankheit sein kann: einige Nahrungszubereitungen
- ( dämpfende Appetit), wie Fenfluramin, Phentermin,
- Einsatz in Lebensmitteln Raps toxisches Öl,
- Empfang anorectics Medikamente( Aminorex Furmat)
- einige Kräutertees Crotalaria Pflanze enthält,
- orale Kontrazeptiva. Symptome
PLG Klinische Manifestationen wie Hypoventilation, Brustschmerzen bei körperlicher Belastung auftreten, können neurotische Störungen erklärt werden. Später durch Atemnot verbunden, Schwäche, Müdigkeit, erscheint Heiserkeit aufgrund der Kompression des Nervus laryngeus aufgrund der erhöhten Lungenarterie, periphere Ödeme, Herzklopfen.
Eine körperliche Untersuchung zeigt eine epigastrische Pulsation durch einen vergrößerten rechten Ventrikel des Herzens, ein galoppierender atrialer Rhythmus ist zu hören, der in den unteren Abschnitten des Sternums nach links besser auditiert wird. Mit der Zerkleinerung wird eine verstärkte Lungenkomponente, rechtsseitiger Galopp oder triguspidales Aufstoßen aufgedeckt. Systolisches Murmeln auf der Pulmonalarterie wird manchmal beobachtet.
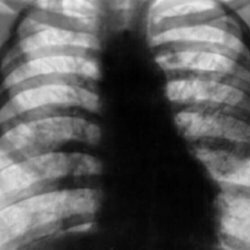
Beim Röntgen der Brust werden die pulmonale Arterienvergrößerung, die rechte ventrikuläre und die rechte atriale Vergrößerung sowie eine scharfe Verengung der Zweige der Pulmonalarterie bestimmt.
Elektrokardiographische Untersuchung zeigt rechtsventrikuläre Überlastung, Achsabweichung nach rechts, Verstärkung von S-Wellen in den unteren Leitungen, R-Wellen größer in Blei V1, Inversion von T-Wellen in rechten Thorax-Leitungen. Der beste Test für die Beobachtung von PLG ist die Doppler-Echokardiographie.
Differenzierung von
Um die korrekte Diagnose von PLG zu etablieren, ist es notwendig, andere bekannte Krankheiten auszuschließen:
- chronische bronchopulmonale Erkrankungen mit pulmonaler Herzbildung;
- Sarkoidose;
- Pneumokoniose( Silikose) - begleitet von hypoxischer pulmonaler Hypertonie;
- angeborene Herzfehler( Herzkatheterisierung und Echokardiographie zur Differenzierung);
- Lungenembolie( erkannt durch Angiopulmonographie oder Radioisotopen der Lunge);
- pulmonale Arterienstenose;
- Eisenmenger-Syndrom, wenn es von einem Defekt des interventrikulären Septums begleitet wird, ähnelt eher PLG;
- Geräusche und galoppierender Rhythmus können zusammen mit PLG ein Zeichen der rheumatischen Aorten- oder Mitralherzkrankheit sein.
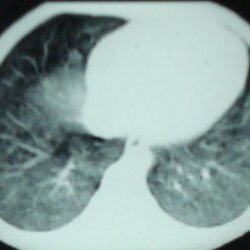
Die informativsten für die Differenzierung und Diagnose der Lungenszintigraphie, Computertomographie und Angiographie. Aus Labortests werden HIV-1-Antikörper, Leber- und Schilddrüsenfunktionstests durchgeführt.
Methoden der Behandlung
Die Behandlung von PLG in den meisten Fällen hat nicht viel Wirkung, ist es möglich, das Leben des Patienten leicht zu erhöhen. In der Regel, trotz der laufenden Therapie, die Krankheit fortschreitet und führt zu einem tödlichen Ergebnis in 2-5 Jahren.
Die Verwendung von Antikoagulantien ist verlängert und wird allen Patienten gezeigt, ohne an PLH zu leiden. Die Verwendung von Medikamenten wie Co-Domadin, Coumadin( Warfarin), Blutverdünnung und Verhinderung der Bildung von Blutgerinnseln, erhöht die Überlebensrate etwas.
Mit reinen ventrikulären Ausfall verschriebenen Herz-Fonds, sowie Diuretika, reduziert peripheren Ödem.
Für leistungsstarke moderne Medikamente, die wirksam sind, aber nicht die Überlebensstatistiken erhöhen, können endogene Vasodilatatoren: Prostaglandin I2( Epoprostenol) zuverlässig eingeschlossen werden. Langzeittherapie mit diesem Medikament kann die Wartezeit für die Transplantation verdoppeln, sowie das Leben des Patienten erhöhen.
Das sicherste Mittel, um den allgemeinen Zustand des Patienten zu entlasten und den Druck zu reduzieren, sind solche Medikamente wie Diazoxid, Nitroglycerin, Nitroprussid, Apressin.
Bei Patienten mit PGD, die Verwendung solcher Medikamente wie:
- b-Agonisten: Isoproterenol, Zatachalin;
- a-adrenoblocker: Phenoxybenzamin, Phentolamin;
- Kalziumkanalblocker - Langzeitbehandlung mit diesen Medikamenten verbessert Hämodynamik und Überleben;
Trotz des hohen Transplantationsrisikos ist die effektivste Behandlungsmethode die Herz- und Lungentransplantation.