
Lipoidosis (Lysosomal lipid storage disease): what is it, examples
Attention! The information is for informational purposes only and is not intended to diagnose and prescribe treatment. Always consult your specialist doctor!
Content
- What are lysosomal storage diseases?
- What are lipids?
- How are lipoidosis inherited?
- Types and symptoms of lysosomal storage diseases
What are lysosomal storage diseases?
Lysosomal storage diseases (lipoidosis), are a group of hereditary metabolic diseases in which harmful amounts of fatty substances (lipids) accumulate in various cells and tissues of the body.
People with these disorders either do not make enough of one of the enzymes needed to break down (metabolize) lipids, or they produce enzymes that do not work properly.
Over time, this excessive accumulation of fat can lead to permanent damage to cells and tissues, especially in the brain, peripheral nervous system (nerves from the spinal cord to the rest of the body), liver, spleen and bone marrow.
What are lipids?
Lipids are fat-like substances that are important parts of the membranes inside and between cells and in the myelin sheath, which covers and protects the nerves. Lipids include oils, fatty acids, waxes, steroids (such as cholesterol and estrogen), and other related compounds.
These fatty substances are naturally stored in the cells, organs and tissues of the body. Tiny bodies within cells, called lysosomes, regularly convert or metabolize lipids and proteins into smaller components to provide energy to the body. Disorders in which intracellular material, which cannot be metabolized, remains in lysosomes, are called lysosomal storage diseases.
In addition to diseases associated with lipid accumulation, other lysosomal diseases associated with accumulation include mucolipidoses, in which cells and tissues accumulate an excessive amount of lipids with sugar molecules attached to them, as well as mucopolysaccharidoses, in which an excessive amount of large, complex sugar accumulates molecules.
How are inherited lipoidosis?
Lysosomal storage diseases are inherited from one or both parents who carry a defective gene that regulates a specific lipid-metabolizing enzyme in a class of cells in the body. They can be inherited in two ways:
- Autosomal recessive inheritance occurs when both parents carry and pass on a copy of the defective gene, but neither parent is affected by the disorder. Every child born to these parents has a 25 percent chance of inheriting both copies of the defective gene, 50 percent the likelihood that he will be a carrier similar to his parents, and a 25 percent chance of not inheriting any copy of the defective gene. Children of either sex can be inherited in an autosomal recessive manner.
- X-linked (or sex-related) recessive inheritance occurs when the mother carries the affected gene on the X chromosome. The X and Y chromosomes are involved in sex determination. Women have two X chromosomes, while men have one X chromosome and one Y chromosome. Sons of female carriers have a 50 percent chance of inheriting and affecting the disorder, as sons receive one X chromosome from their mother and one Y chromosome from their father. Daughters have a 50 percent chance of inheriting the affected X chromosome from their mother and are carriers or mildly affected. Affected men do not transmit the disease to their sons, but their daughters will be carriers and carriers of the disease.
Types and symptoms of lysosomal storage diseases
Gaucher disease - the most common of lysosomal diseases. The disease is caused by a deficiency of the enzyme glucocerebrosidase, which leads to the accumulation of glucocerebroside in many tissues. Fatty material can accumulate in the brain, spleen, liver, kidneys, lungs, and bone marrow.
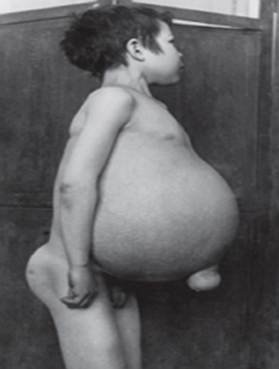
Symptoms may include brain damage, enlargement of the spleen and liver, impaired liver function, skeletal disorders and bone damage that can cause pain and fractures, swelling of the lymph nodes and (sometimes) adjacent joints, bloating, brownish skin tone, anemia, low platelets and yellow spots on the eyes.
The individuals most seriously affected may also be more susceptible to infections. The disease affects men and women equally.
Gaucher disease has three general clinical subtypes:
- Type 1 (Or no neuronopathic type) is the most common form of the disease in the United States and Europe. The brain is not affected, but there may be damage to the lungs and, in rare cases, the kidneys. Symptoms can begin early in life or into adulthood and include an enlarged liver and severe enlargement of the spleen, which can lead to rupture and additional complications. Skeletal weakness and bone disease can be extensive. People in this group usually bruise easily due to the low platelet count in their blood. They may also experience fatigue due to anemia. Depending on the onset and severity of the disease, people with type 1 can live to adulthood. Many sufferers have mild illness or may not show any symptoms. Though 1type Gaucher disease is common among people of Ashkenazi Jewish heritage and can affect people of all ethnic backgrounds.
- Type 2 (or acute childhood neuropathic Gaucher disease) usually begins within 3 months of birth. Symptoms include extensive and progressive brain damage, spasticity, seizures, stiff limbs, enlarged liver (liver hepatomegaly) and spleen (splenomegaly), abnormal eye movement, and poor ability to suck and swallow. Affected children usually die before 2 years of age.
- Type 3 (chronic neuronopathic form) can begin at any time during childhood or even in adulthood. It is characterized by slowly progressive but less severe neurological symptoms compared to acute Gaucher disease 2 types. Major symptoms include eye movement disorders, cognitive deficits, poor coordination, seizures, enlargement of the spleen and / or liver, skeletal disorders, blood disorders including anemia, and problems with breathing. Almost everyone with Gaucher disease 3 types, who receives enzyme replacement therapy will reach adulthood.
For patients 1st and 3rd types Enzyme replacement therapy, given intravenously every two weeks, can dramatically reduce the size of the liver and spleen, reduce skeletal abnormalities, and reverse other manifestations. A successful bone marrow transplant treats the neurological manifestations of the disease. However, this procedure carries significant risks and is rarely done in individuals with Gaucher disease.
In rare cases, surgery may be required to remove all or part of the spleen (if the person has a very low platelet count or when an enlarged organ greatly affects the person's comfort). Blood transfusion may benefit some people with anemia. Others may need joint replacement surgery to improve mobility and quality of life. There is currently no effective treatment for the brain damage that can occur in people with type 2 and 3 Gaucher disease.
Niemann-Pick disease is a group of autosomal recessive disorders caused by the accumulation of fat and cholesterol in the cells of the liver, spleen, bone marrow, lungs and, in some cases, the brain.
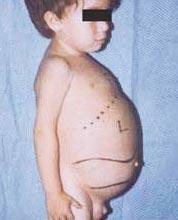
Neurological complications can include ataxia (lack of muscle coordination that can affect walking, writing and eating, among other functions), eye paralysis, brain degeneration, learning problems, spasticity, difficulty feeding and swallowing, slurred speech, loss of muscle tone, hypersensitivity to touch, and some corneal opacity due to excessive accumulation materials. In 50% of affected people, a characteristic cherry-red halo develops around the center of the retina, which can be seen by a doctor with a special instrument.
Niemann-Pick disease is classified into three general subtypes:
- Type A, the most severe form, begins in early childhood. Babies appear normal at birth, but by 6 months, deep brain damage develops, an enlarged liver and spleen, and swollen lymph nodes and nodes under the skin (xanthomas). The spleen may become 10 times its normal size and may rupture, causing bleeding. These children become increasingly weak, lose motor function, may become anemic, and are prone to recurrent infections. They rarely live more than 18 months. This form of the disease is most common in Jewish families.
- Type B(or juvenile onset) usually does not affect the brain, but most children develop ataxia, damage to the nerves leaving the spinal cord (peripheral neuropathy), and pulmonary difficulties that progress with age. Enlargement of the liver and spleen is typical for adolescents. People with type B can live relatively long, but many require lifelong oxygen therapy due to lung damage. Niemann-Pick types A and B are the result of an accumulation of a fatty substance called sphingomyelin due to a deficiency in an enzyme called sphingomyelinase.
- Type C may appear early in life or develop during adolescence or even adulthood. Niemann-Pick diseasetype C is caused not by sphlingomyelinase deficiency, but by a lack of NPC1 or NPC2 proteins. As a result, various lipids and especially cholesterol accumulate in nerve cells and cause their malfunction. Brain involvement can be extensive, resulting in inability to look up and down, difficulty walking and swallowing, progressive hearing loss, and progressive dementia. People with type C have only mild enlargement of the spleen and liver. Life expectancy of people with type C varies considerably. Some people die in childhood, while others who are less affected may also live in adulthood.
There is currently no cure for Niemann-Pick disease. Treatment is supportive. Children usually die from infection or progressive neurological loss. There have been cases of bone marrow transplantation in several patients with type B with mixed results.
Fabry diseasealso known as alpha-galactosidase-A deficiency, causes the accumulation of fatty matter in the autonomic nervous system (that part of the nervous system that controls involuntary functions such as breathing and heartbeat), in the eyes, kidneys and cardiovascular system.
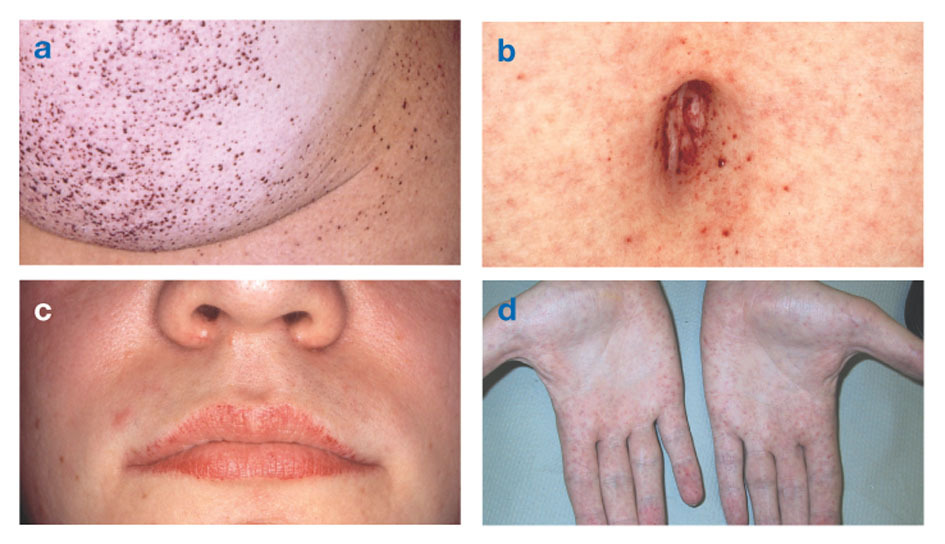
Fabry disease is the only X-linked lipid disease. The disease mainly affects men, although a milder and more variable form occurs in women. Rarely, affected women have severe symptoms similar to those seen in men with the disorder. The onset of symptoms usually occurs during childhood or adolescence.
Neurologic signs include burning pain in the arms and legs that gets worse in hot weather or after exercise, as well as the accumulation of excess material in the transparent layers of the cornea (which leads to clouding, but without changes in vision). Fat accumulation in the walls of blood vessels can disrupt circulation, putting a person at risk stroke or heart attack. Other symptoms include an enlarged heart, progressive renal failureleading to end-stage renal disease, gastrointestinal problems, decreased sweating and fever.
Angiokeratomas (small, non-cancerous, reddish-purple raised patches on the skin) can develop in the lower torso and become more common with age.
People with Fabry disease often die prematurely from complications from heart failure, kidney failure, or stroke. Medicines such as phenytoin and carbamazepine are often prescribed to treat the pain that accompanies Fabry disease, but not to cure it. Metoclopramide or Lipisorb (dietary supplement) may relieve gastrointestinal upset that often occurs in people with Fabry disease, and some people may need a kidney transplant or dialysis. Enzyme replacement can reduce storage, relieve pain, and preserve organ function in some people with Fabry disease.
Farber's disease also known as Farber's lipogranulomatosis, describes a group of rare autosomal recessive disorders that cause the accumulation of fatty matter in joints, tissues, and the central nervous system. It affects both men and women. The disease usually begins in early childhood, but can occur later in life.
Children with classic Farber's disease develop neurological symptoms, which may include increased sleepiness and lethargy, and problems with swallowing. The liver, heart, and kidneys can also be affected. Other symptoms may include joint contractures (chronic shortening of muscles or tendons around the joints), vomiting, arthritis, enlargement lymph nodes, joint inflammation, hoarseness, and lumps under the skin that thicken around the joints as it progresses diseases.
Affected people with breathing difficulties may need to have a breathing tube inserted. Most children with this condition die by age 2, usually from lung diseases. In one of the most severe forms of the disease, an enlarged liver and spleen can be diagnosed shortly after birth. Babies born with this form of the disease usually die within 6 months.
Farber's disease is caused by a deficiency of an enzyme called ceramidase. There is currently no specific treatment for Farber's disease. Corticosteroids may be prescribed to relieve pain. Bone marrow transplantation can reduce granulomas (small masses of inflamed tissue) in people with mild inflammation or in patients without pulmonary or nervous system complications. In older people, granulomas can be shrunk or surgically removed.
Gangliosidosis consists of two different groups of genetic diseases. Both are autosomal recessive and affect males and females equally.
GM1 gangliosidosis.
GM1 gangliosidosis is caused by a deficiency of the enzyme beta-galactosidase, resulting in an abnormal accumulation of acidic lipid materials, in particular in nerve cells in the central and peripheral nervous systems. GM1 gangliosidosis has three clinical manifestations:
- GM1 (the most severe subtype, occurring soon after birth) may include neurodegeneration, seizures, enlarged liver and spleen, coarseness of facial features, skeletal irregularities, joint stiffness, bloating, muscle weakness, exaggerated startle response, and problems with gait. About half of those affected develop cherry-red spots in the eye. Children may be deaf and blind by age 1 and often die by age 3 from heart complications or pneumonia.
- Late child GM1 gangliosidosis usually begins between the ages of 1 and 3 years. Neurologic symptoms include ataxia, seizures, dementia, and difficulty speaking.
- GM1 gangliosidosis develops between the ages of 3 and 30 years. Symptoms include decreased muscle mass (muscle atrophy), neurological complications that are less severe and progress more slowly than other forms disorders, corneal opacity in some people, and sustained muscle contractions that cause twisting and repetitive movements or abnormal postures (dystonia). Angiokeratomas can develop in the lower trunk of the body. The size of the liver and spleen is normal in most affected people.
GM2 gangliosidosis.
GM2 gangliosidosis also causes the body to accumulate excess acidic fatty substances in tissues and cells, primarily nerve cells. These disorders result from a deficiency of the enzyme beta-hexosaminidase. GM2 disorders include:
- Tay-Sachs disease (also known as GM2 gangliosidosis variant B) and its variant forms are caused by a deficiency of the enzyme hexosaminidase A. Affected children develop normally during the first few months of life. Symptoms begin at 6 months of age and include progressive loss of mental capacity, dementia, decreased eye contact, increased a startle response to noise, progressive hearing loss leading to deafness, difficulty swallowing, blindness, cherry red spots on the retina, and some paralysis. Seizures can begin in the second year of a child's life. Babies may eventually become dependent on the feeding tube, and often die by the age of 4 from recurring infections. Unfortunately, there is no special treatment. Anticonvulsants may initially control seizures. Other supportive treatments include proper nutrition and hydration, and methods for keeping the airways open. A rare form of the disorder called late-onset Tay-Sachs disease, occurs in people between the ages of 20 and 30 and is characterized by an unstable gait and progressive neurological deterioration.
- Sandhoff's disease (Option AB) is a severe form of Tay-Sachs disease. Onset usually occurs at 6 months of age and is not restricted to any ethnic group. Neurological signs may include progressive deterioration of the central nervous system, motor weakness, early blindness, severe frightened reaction to sound, spasticity, shock, or twitching muscles (myoclonus), epilepsy, an abnormally enlarged head (macrocephaly), and red patches in the eye. Other symptoms may include frequent respiratory infections, heart murmur, doll-like facial features and enlargement of the liver and spleen. There is no specific treatment for Sandhoff disease. As with Tay-Sachs disease, supportive care includes keeping the airways open at all times, as well as proper nutrition and hydration. Anticonvulsants can initially control epilepsy (seizures).
Krabbe's disease(also known as globular cell leukodystrophy and galactosylceramide lipidosis) is an autosomal recessive disease caused by a deficiency of the enzyme galactocerebrosidase. The disease most commonly affects infants with onset before 6 months of age, but can occur during adolescence or adulthood.
The accumulation of undigested fats affects the growth of the nerve's protective sheathing (myelin sheath) and causes severe impairment of mental and motor skills. Other symptoms include muscle weakness, decreased ability of the muscles to stretch (hypotonia), muscle tension (spasticity), sudden shock or twitching of the limbs (myoclonic seizures), irritability, unexplained fever, deafness, blindness, paralysis, and difficulty swallowing. Long-term weight loss can also occur.
The disease can be diagnosed by enzyme testing and identifying its characteristic cell grouping into globoid bodies in the white matter of the brain, demyelination of nerves and degeneration, and destruction of brain cells. In infants, the disease is usually fatal before 2 years of age. People with a more advanced form of the disease have a milder course of the disease and live significantly longer.
No specific treatment for Krabbe's disease has been developed, although early bone marrow transplantation may help some people. People with a more advanced form of the disease have a milder course of the disease and live significantly longer.
Metachromatic leukodystrophy, or MLD, is a group of disorders marked by the accumulation of fatty substances in the white matter of the central nervous system and in peripheral nerves and to some extent in the kidneys. Similar to Krabbe's disease, MLD affects the myelin, which covers and protects the nerves. This autosomal recessive disorder is caused by a deficiency of the enzyme arylsulfatase A. This disorder affects both men and women.
Metachromatic leukodystrophy has three characteristic forms: late infant, juvenile, and adult.
- Late Infant MLD usually starts between 12 and 20 months after birth. Children may appear normal at first, but have difficulty walking and a tendency to fall, accompanied by intermittent pain in the arms and legs. progressive loss of vision leading to blindness, developmental delays and loss of previously acquired milestones, impaired swallowing, seizures, and dementia up to 2 years. Children also develop progressive muscle wasting and weakness and eventually lose their ability to walk. Most children with this form of the disorder die by age 5.
- Juvenile MLD usually begins between the ages of 3 and 10 years. Symptoms include impaired school performance, mental impairment, ataxia, seizures, and dementia. Symptoms progress, with death occurring 10–20 years after the onset of the disease.
- Adult form. Symptoms adults onset after age 16 and may include ataxia, seizures, abnormal tremors of the limbs (tremors), impaired concentration, depression, mental disorders and dementia. Death usually occurs 6–14 years after the onset of symptoms.
MLD cannot be cured. Treatment is symptomatic and supportive. Bone marrow transplantation can in some cases delay the progression of the disease. Significant progress has been made with gene therapy in animal models with MLD and in clinical trials.
Wolman's diseasealso known as lysosomal acid lipase deficiencyis a serious lipid storage disorder that is usually fatal at 1 year of age. This autosomal recessive disorder is characterized by an accumulation of cholesterol esters (usually the transport form of cholesterol) and triglycerides (the chemical form in which fats exist in the body), which can accumulate significantly and cause cell damage and fabrics.
Both men and women suffer from this disorder. Infants are normal and active at birth, but rapidly develop progressive mental impairment, an enlarged liver and greatly enlarged spleen, bloating, gastrointestinal problems. jaundice, anemia, vomiting and calcium deposits in adrenal glandscausing them to harden.
Another type lysosomal acid lipase deficiency is cholesterol ester storage disease. This extremely rare disease occurs as a result of the storage of cholesterol esters and triglycerides in blood cells, lymph and lymphoid tissue. In children in adulthood, the liver enlarges, which leads to cirrhosis and chronic liver failure. Children may also have calcium deposits in the adrenal glands, and jaundice may develop in the later stages of the disease.
Currently, the issue of enzyme replacement is being actively studied both in Wolman's disease and in the accumulation of a cholesterol ester.
Attention! The information is for informational purposes only and is not intended to diagnose and prescribe treatment. Always consult your specialist doctor!

