
Pheochromocytoma: Symptoms, Diagnosis and Treatment
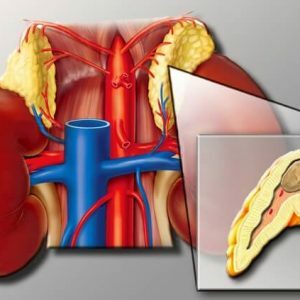 Pheochromocytoma( chromaffinoma) is a tumor that is formed by the pathological proliferation of chromaffin cells and is characterized by hormonal activity.It most often comes from the adrenal medulla, and can be both benign and malignant.
Pheochromocytoma( chromaffinoma) is a tumor that is formed by the pathological proliferation of chromaffin cells and is characterized by hormonal activity.It most often comes from the adrenal medulla, and can be both benign and malignant.
Pheochromocytomas often develop in young and adulthood( 20-40 years), with prevalence among men and women being approximately the same as .In childhood, the incidence is higher among boys.The tumor produces an excessive amount of peptides and biogenic amines( dopamine, epinephrine and norepinephrine), resulting in catecholamine crises.
Table of Contents: Etiology Pathogenesis Symptoms of adrenal pheochromocytoma Diagnosis of pheochromocytoma Treatment and prognosisMalignant tumor types( pheochromoblast) account for less than 10% of the total number diagnosed with pheochromocytomas. For such tumors, the location outside the adrenal gland is very characteristic.Secondary foci( metastases) are formed in regional lymph nodes, liver, distant organs( lungs) and tissues( muscular and bone).
Etiology
Generally, the true cause of chromaffin formation remains unclarified.
Every tenth patient has a genetic predisposition for .During the collection of the anamnesis, it turns out that these tumors were previously diagnosed in the parents.Genetics believe that pathology is inherited by an autosomal dominant type.
Often pheochromocytoma is one of the manifestations of a hereditary disease - the syndrome of multiple endocrine neoplasias .When it affects other organs of the endocrine system - the thyroid and parathyroid glands.
Pathogenesis of
A pheochromocytoma can occur not only from the adrenal medulla but also from the aortic lumbar paraganglia( in such cases, they are referred to as paragangliomas).Neoplasms of this type are also revealed in the area of the small pelvis, thoracic and abdominal cavity.In rare cases, chromaffinoma is found in the head and neck area.The cases of tumor localization in the pericardium and heart muscle are described.
Active substances that can be synthesized by chromaffinoma:
-
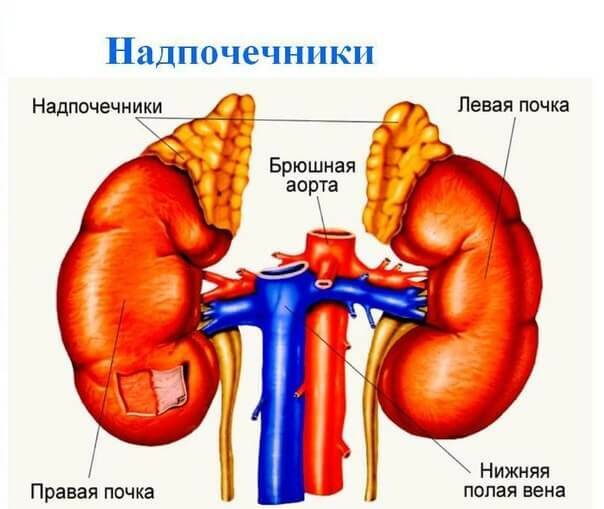 dopamine;
dopamine; - adrenaline;
- noradrenaline;
- calcitonin;
- Somatostatin;
- adrenocorticotropic hormone;
- serotonin;
- is a vasoactive intestinal polypeptide.
The pheochromocytoma is capable of producing the neuropeptide Y, which is characterized by pronounced vasoconstrictor properties.
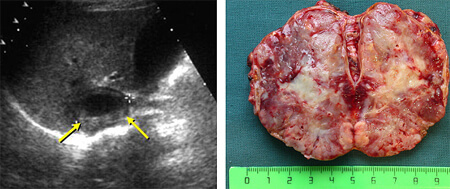
The level of hormonal activity does not depend on the size of the lesion( they are variable, and can reach 5 cm).The average weight of pheochromocytoma is 70 g. For these encapsulated tumors, rich vascularization is characteristic.
Symptoms of adrenal pheochromocytoma
 Symptoms are determined by the excess of a substance .Hyper secretion of catecholamines with pheochromocytoma leads to the development of arterial hypertension.Tumors are detected in approximately one hundredth patient with constantly increased diastolic( "lower") pressure.
Symptoms are determined by the excess of a substance .Hyper secretion of catecholamines with pheochromocytoma leads to the development of arterial hypertension.Tumors are detected in approximately one hundredth patient with constantly increased diastolic( "lower") pressure.
The course of hypertension can be stable or paroxysmal.Periodic crises are accompanied by violations from the cardiovascular and nervous system, as well as digestive and metabolic disorders.
During a crisis, blood pressure rises sharply, and at intervals between paroxysms it is stably high or returns to normal values.
Symptoms of crisis with pheochromocytoma:
- increase in blood pressure to 200 mm.Gt;Art.and more;
- unmotivated feeling of anxiety and fear;
- intense headaches;
- tremor;
- chills;
- skin blushing;
- hyperhidrosis( increased sweating);
- cardialgia( pain in the region of the heart);
- heart palpitations;
- arrhythmia;
- convulsions;
- dyspepsia disorder.
During an attack in peripheral blood, there is an increased level of sugar and leukocytosis.
The duration of the paroxysm is from a few minutes to an hour or more.Their frequency varies from single seizures for several months to 10-15 per day. Crisis is characterized by a sharp spontaneous cupping, which is accompanied by a sharp drop in blood pressure.The patient is noted for pouring sweat and increased urine( up to 5 L) with a low specific gravity.He complains of general weakness and a sense of "brokenness" in the whole body.
Factors capable of provoking a crisis:
- general overheating or subcooling of the body;
- considerable physical activity;
- psychoemotional stresses;
- reception of some pharmacological agents;
- Alcohol consumption;
- sharp movements;
- medical manipulation( deep palpation of the abdomen).
 The most severe outcome of an attack is catecholamine shock.It is characterized by uncontrolled hemodynamics - episodes of hypotension and hypertension are randomly replaced and do not lend themselves to medical correction. In severe hypertensive crisis caused by chromaffinoma, such complications as pulmonary edema, stroke, infarction, exfoliating aortic aneurysm, development of functional kidney failure are not ruled out.Frequent hemorrhages in the retina. The greatest danger of paroxysms is for women during pregnancy.
The most severe outcome of an attack is catecholamine shock.It is characterized by uncontrolled hemodynamics - episodes of hypotension and hypertension are randomly replaced and do not lend themselves to medical correction. In severe hypertensive crisis caused by chromaffinoma, such complications as pulmonary edema, stroke, infarction, exfoliating aortic aneurysm, development of functional kidney failure are not ruled out.Frequent hemorrhages in the retina. The greatest danger of paroxysms is for women during pregnancy.
With stable flow, the patient has a stable high blood pressure, against which the pathologies of the heart muscle and kidneys develop over time, as well as changes in the fundus of the .For patients with pheochromocytoma, lability of the psyche( mood swings and high psychoemotional excitability), periodic cephalgia and increased physical and mental fatigue are characteristic.
Among the metabolic disorders in particular is the increase in blood glucose( hyperglycemia), which often causes the development of diabetes.
Important: with pheochromoblastomas( malignant chromaffinomas), the patient has cachexia( a sharp decrease in body weight) and pains the abdominal area.
Diagnostics of pheochromocytoma
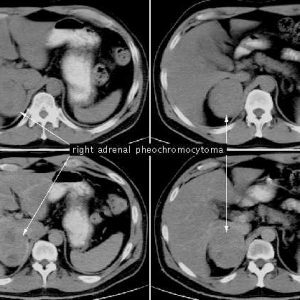 In the course of the general examination, patients experience palpitations, pale skin of the face, neck and chest, increased blood pressure. Orthostatic hypotension is also characteristic( when a person rises, the pressure drops sharply).
In the course of the general examination, patients experience palpitations, pale skin of the face, neck and chest, increased blood pressure. Orthostatic hypotension is also characteristic( when a person rises, the pressure drops sharply).
Important: palpation of the neoplasm can provoke catecholamine paroxysm.
One of the important diagnostic criteria is an increase in the content of catecholamines in the urine and blood of the .In the serum, also determine the level of chromogranin-A( universal transport protein), adrenocorticotropic hormone, calcitonin and trace elements - calcium and phosphorus.
Nonspecific changes in the electrocardiogram are usually determined only during the crisis period.
Pheochromacytomas often have accompanying pathologies - cholelithiasis, neurofibromatosis, arterial blood flow disorders in the extremities( Raynaud's syndrome) and hypercorticism with the development of the Itenko-Cushing syndrome.
A significant proportion of the subjects found hypertension-mediated retinal vascular lesions( retinopathy). All patients with suspected pheochromocytoma need to undergo an additional examination from an ophthalmologist.
In the differential diagnosis of chromaffin, provocative( stimulating) and suppressive tests with histamine and tropafene are used, but it is possible to obtain false positive and false negative results of the samples.
Of the hardware diagnostic methods, ultrasound scanning and tomographic examination( CT and MRI) of the adrenal glands are considered the most informative. They allow you to specify the size and location of the lesion.In addition, selective arteriography and adrenal scintigraphy, as well as radiographic examination of the chest( to confirm or exclude the intrathoracic localization of chromaffinoma) are resorted to.
Pathology with which differential diagnosis is performed:
- thyrotoxicosis;
- neuroses;
- psychosis;
- encephalitis;
- craniocerebral trauma;
- strokes;
- transient ischemia of the brain;
- paroxysmal tachycardia;
- hypertension;
- some types of poisoning.
Note: in pregnant women, the symptomatology of pheochromocytoma is masked for late toxicosis( gestosis) and the most severe forms of their course are eclampsia and preeclampsia.
It is not always possible to establish the malignancy of a tumor at the preoperative stage reliably. It is possible to speak with confidence about pheochromoblastoma if there are such obvious signs as invasion( germination) in nearby structures or distant secondary foci.
Treatment and prognosis
 When diagnosed with pheochromocytomas, medication is used to reduce the severity of clinical symptoms and stop the paroxysmal attacks of .Conservative measures suggest the administration of drugs from the group of α-adrenoblockers( Fentolamine, Phenoxybenzamine, Tropafen, and in the preoperative period - Doxazosin) and β-blockers( Metoprolol, Propranolol).In the case of crises, nitroprusside sodium is additionally introduced.A very effective drug for lowering the level of catecholamines is A-methyltyrosine, but its regular intake can provoke mental disorders and digestive disorders.
When diagnosed with pheochromocytomas, medication is used to reduce the severity of clinical symptoms and stop the paroxysmal attacks of .Conservative measures suggest the administration of drugs from the group of α-adrenoblockers( Fentolamine, Phenoxybenzamine, Tropafen, and in the preoperative period - Doxazosin) and β-blockers( Metoprolol, Propranolol).In the case of crises, nitroprusside sodium is additionally introduced.A very effective drug for lowering the level of catecholamines is A-methyltyrosine, but its regular intake can provoke mental disorders and digestive disorders.
Surgery is then performed - total adrenalectomy.During surgery, the affected adrenal gland is removed along with the tumor.Since the probability of the location of pheochromocytoma outside the adrenal gland and the presence of multiple neoplasms is high, a "classical" laparotomy approach is preferred, but a less traumatic laparoscopic intervention is also possible.
Multiple endocrine neoplasia is an indication for resection of both adrenal glands.
When diagnosing pheochromocytoma in a pregnant patient, depending on the term, an artificial interruption or cesarean section is performed, and then - removal of the tumor.
In establishing the malignant nature of the neoplasm and the detection of distant secondary foci, chemotherapy is indicated.Such patients need course treatment with cytostatic drugs - Dacarbazine, Vincristine or Cyclophosphamide.
In most cases, after resection of a benign neoplasm, blood pressure values are normalized, and other clinical signs regress .If hypertension persists, there is reason to assume the presence of ectopic tumor tissue, incomplete removal or accidental damage to the renal artery.
A single standard for surgical interventions for multiple tumors has not been developed.Sometimes it is advisable to carry out a resection in several stages.
Five-year survival rates after surgery for benign chromaffinoma are 95%.After resection of pheochromoblastoma, the prognosis is less favorable.
These hormone-active tumors tend to recur about 12% of the time.All patients who underwent surgery are strongly recommended to be examined annually by an endocrinologist.
Vladimir Plisov, medical reviewer
