
Robinovi sündroom: mis see on, põhjused, nähud, ravi, prognoos
Sisu
- Mis on Robinoffi sündroom?
- Robinovi sündroomi kliinilised tunnused
- Robinovi sündroomi põhjused
- Mõjutatud populatsioonid
- Diagnostika
- Robinovi sündroomi ravi
- Prognoos
- Sarnased häired
Mis on Robinoffi sündroom?
Robinoffi sündroom on äärmiselt haruldane pärilik haigus, mis mõjutab luude ja teiste kehaosade arengut. Robinovi sündroomil on kaks vormi, mis erinevad tunnuste ja sümptomite, raskusastme, pärandi tüübi ja nendega seotud geenide poolest.
Esiteks, autosoomne retsessiivne Robinovi sündroom on raskem ja seda iseloomustavad:
- käte ja jalgade pikkade luude lühendamine;
- lühikesed sõrmed ja varbad;
- selgroo selgroolised luud, mis põhjustab selgroo ebanormaalset kõverust (kyphoscoliosis);
- ribide tühjendamine või puudumine;
- lühike kasv;
- iseloomulikud näojooned.
Muud funktsioonid võivad hõlmata järgmist:
- suguelundite vähearenemine;
- hambaprobleemid;
- neeru- või südamepuudulikkus;
- arengu viivitus.
Lapsed koos teine vorm, autosomaalselt domineeriva Robinovi sündroomi sümptomid on sarnased, kuid avalduvad leebemalt. Lülisamba ja ribide anomaaliaid tavaliselt ei esine ning lühike kasv on vähem väljendunud.
Mõnedel autosomaalse domineeriva Robinovi sündroomiga inimestel võib samuti olla suurenenud luude mineraalne tihedus (osteoskleroos).
Robinovi sündroomi kliinilised tunnused

Robinovi autosomaalset retsessiivset sündroomi iseloomustavad:
- lühike kasv;
- iseloomulikud näojooned;
- luustiku ja / või suguelundite kõrvalekalded.
Kliiniliste tunnuste ulatus ja raskusaste on inimestel erinev.
Enamik Robinoffi sündroomiga lapsi kogeb kasvupeetus pärast sündimille tulemuseks on väike või mõõdukalt lühike kasv. Enamikul lastest on normaalne intelligentsus, kuid umbes 20 protsendil kannatanutest võib olla:
- vaimne kahjustus;
- viivitused verstapostide saavutamisel;
- viivitused keeleoskuse arendamisel.
Näo omadused sarnanevad areneva loote tunnusedMeditsiinikirjanduses nimetatakse seda sageli loote näoks. Pea ja näopiirkonna iseloomulikud kõrvalekalded võivad hõlmata suur pea (makrotsefaalia) punnis laubaga ja keskpinna vähearenenud (keskpinna hüpoplaasia).
Mõjutatud lastel võib olla ka:
- Laiade vahedega silmad (silma hüpertelorism), mis ulatuvad orbiitidest välja (punnis silmad)
- ebatavaliselt laiad kaldus silmalaud (silmade pilud);
- väike ülespoole pööratud nina, mille ninasõõrmed laienevad ettepoole;
- uppunud vibusild;
- Valesti asetatud (st madalal asetsevad, tagasi pööratud) kõrvad.
Mõnel mõjutatud lapsel võib olla:
- lai, kolmnurkne suu, pööratud allapoole, ülahuule keskel pikk pilu;
- väike lõug;
- väike lõualuu (mikrognaatia);
- igemete hüperplaasia.
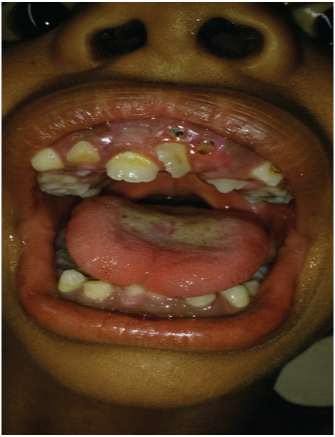
Võib esineda ka hammaste kõrvalekaldeid, sealhulgas:
- hammaste ebaühtlane asend;
- hammaste tagumine kokkutõmbumine;
- sekundaarsete (püsivate) hammaste hilinenud purskamine.
Pehmete kudede struktuur kurgu tagaosas (uvula) võib olla vähearenenud või ebanormaalselt jagunenud (kaheharuline). Mõjutatud lastel võib olla ka suulaelõhe, ebanormaalne vertikaalne soon või ülahuule auk (huulelõhe) ja / või keele piiratud liikumine (anküloglossia).
Ankyloglossia võib kaasa aidata keeleoskuse arengu viivitustele. Enamikul lastel muutuvad Robinovi sündroomiga seotud näohäired vanusega vähem väljendunud.
Skeleti kõrvalekalded võivad hõlmata järgmist:
- küünarvarre luude (raadius ja küünarluu) patoloogiad, mis näevad välja ebanormaalselt lühikesed ja vähearenenud (küünarvarre brahhüelia);
- küünarvarre pöidla külje ebatavaline kõrvalekalle (raadius) raadiuse lühendamise (madelung randme) tõttu;
- ebatavaliselt lühikesed sõrmed (brachydactyly);
- viienda varba püsiv fikseerimine painutatud asendis (klinodactyly);
- ebanormaalselt väikesed käed laiade pöidlatega.
Suurte varvaste terminaalsed luud (terminaalsed falangid) võivad olla ebanormaalselt eraldatud ja / või sõrmede ja varvaste luud (falangid) võivad olla vähearenenud (hüpoplastilised). Täiendavad kõrvalekalded võivad hõlmata järgmist:
- puusaliigese nihkumine;
- piiratud küünarnuki pikendamine, ebanormaalne sulandumine või teatud ribide puudumine;
- selgroo ebanormaalne kõverus küljelt küljele (skolioos);
- luude (selgroolülide) ühe külje vähearenemine selgroo keskosas (rindkere) (hemivertebra);
- mõnede selgroolülide liitmine.
Loe ka:Ehlers-Danlose sündroom
Mõjutatud lapsed võivad kogeda luude ebanormaalne süveneminemoodustades rinna keskosa ("lehtri rind"). Lastel võivad olla deformeerunud (düsplastilised) küüned ja / või kõrvalekalded sõrmede ja peopesade nahaharja mustrites.
Enamikul Robinoffi sündroomiga lastel on ka suguelundite anomaaliadja mõnel võivad olla välised suguelundid, mis ei ole selgelt mehed ega naised (mitmetähenduslikud suguelundid). Tavaliselt saab soo õigesti kindlaks teha varases lapsepõlves. Naiste seas Kliitor ja naha pikemad voldid mõlemal pool tupeava (labia majora) võivad olla vähearenenud (hüpoplastilised). Meestel peenis võib olla ebanormaalselt väike (mikropenis) ja peidetud ümbritseva naha alla; Lisaks ei pruugi üks või mõlemad munandid laskuda munandikotti (krüptorhidism).
Harva võivad mõjutatud meestel olla ebanormaalselt madalad munandite funktsioonid (osaline esmane) hüpogonadism, kuid sekundaarsete seksuaalomaduste normaalne areng (näiteks jäme hääl, iseloomulikud mustrid) juuste kasv, munandite ja munandikoti kasvu ja arengu järsk suurenemine jne), välja arvatud püsivus mikropenis.
Mõjutatud naised näitavad normaalset munasarjade funktsiooni (sugunäärmete funktsiooni) ja normaalset viljakust.
Robinoffi sündroomiga inimestel võib olla täiendavaid füüsilisi puudeid, näiteks:
- neerude kahekordistumine;
- ebatavaline uriini kogunemine neerudesse (hüdroonefroos);
- käärsoole osade punnis kõhu limaskesta ebanormaalse ava kaudu (kubemesong);
- käärsoole osade punnitamine läbi kõhuseina naba lähedal (nabasong).
Lisaks on umbes 13 protsenti Robinoffi sündroomiga beebidest sündinud südame defektid (kaasasündinud südamepuudulikkus). Sellistel juhtudel on kõige sagedasem südamepuudulikkus "parema vatsakese obstruktsioon", mille korral verevool südame alumisest kambrist (vatsakesest) on blokeeritud. veresoone ebanormaalse kitsenemise (stenoos) või sulgemise (atresia) tõttu, mis tuleneb vatsakesest (kopsutüvi) ja jaguneb vasakuks ja paremaks kopsuarteriks arterid.
Selle südamepuudulikkusega seotud sümptomid varieeruvad sõltuvalt obstruktsiooni suurusest ja asukohast. Mõnedel Robinoffi sündroomiga lastel võivad olla muud südameprobleemid, sealhulgas keerulised kaasasündinud südamerikked, mis võivad põhjustada eluohtlikke tüsistusi.
Harva võivad Robinoffi sündroomiga imikud ja lapsed olla altid korduvatele kopsuinfektsioonidele (kopsupõletik). Harvadel ja rasketel juhtudel ilma sobiva ravita kopsupõletik võib põhjustada eluohtlikke tüsistusi.
Robinovi sündroomi domineerivatel ja retsessiivsetel vormidel on palju samu sümptomeid ja füüsilisi nähud (nt näonäo kõrvalekalded, lühike kasv, luustiku väärarengud ja suguelundite hüpoplaasia elundid). Kuid retsessiivse vormiga seotud sümptomid ja füüsilised tunnused kipuvad olema raskemad.
Retsessiivse Robinovi sündroomiga imikutel ilmneb roiete arvulisi kõrvalekaldeid (nt ebanormaalne nihe, sulandumine) ja / või teatud ribide puudumine) ja lülisamba (selgroolülide) luid mõjutavad defektid kui domineerivatel imikutel haigused. Lisaks on tõsisem lühike kasv, küünarvarre luude vähearenenud areng (kiirgushüpoplaasia) ja sõrmede kõrvalekalded. Mõjutatud lastel võib olla küünarvarre ühe luu pea nihestus ja seda anomaaliat täheldatakse harva Robinovi sündroomi domineeriva vormiga inimestel. Retsessiivse vormiga inimestel võib olla ka kolmnurkne suu kuju.
Autosomaalse domineeriva Robinovi sündroomi varianti, osteosklerootilist vormi, iseloomustab:
- suurenenud luude mineraalne tihedus, eriti koljus;
- normaalne kasv;
- suur pea;
- ja kuulmislangus, lisaks tüüpilistele tunnustele ja sümptomitele.
Loe ka:Atsetoon lapse uriinis (atsetonuuria lastel): mida teha, põhjused, sümptomid, ravi
Robinovi sündroomi põhjused

Robini sündroomi autosoomsed retsessiivsed ja autosomaalsed domineerivad vormid on põhjustatud muutustest (mutatsioonidest) erinevates geenides.
Autosomaalne retsessiivne tüüp Haigus esineb geeni mutatsioonide tõttu ROR2mis põhjustab valgu puudust ROR2. Selle valgu puudumisel on lapse areng häiritud, eriti luustiku, südame ja suguelundite moodustumine.
Autosomaalne domineeriv tüüp Haigus esineb geeni mutatsioonide tõttu WNT5A või geen DVL1, mis põhjustab normaalseks arenguks vajalike spetsiifiliste valkude puudust. Osteosklerootilise vormi põhjuseks on mutatsioonid geen DVL1.
Väikesel protsendil Robinoffi sündroomiga lastest ei esine üheski neist geenidest mutatsioone ning haiguse põhjus jääb teadmata.
Enamiku geneetiliste haiguste määrab geeni kahe koopia staatus, üks isalt ja teine emalt. Retsessiivsed geneetilised häired tekivad siis, kui inimene pärib sama tunnuse ebanormaalse geeni kaks eksemplari, ühe mõlemalt vanemalt. Kui inimene pärib haiguse jaoks ühe normaalse geeni ja ühe geeni, kannab ta seda haigust, kuid tavaliselt ei näita ta mingeid sümptomeid. Kahe kandjavanema risk, kes mõlemad muudetud geeni edasi kannavad ja last nakatavad, on iga raseduse korral 25%. Lapse saamise oht, kes saab kandjaks, nagu vanem, on iga raseduse korral 50%. Lapsel on 25% tõenäosus saada normaalseid geene mõlemalt vanemalt. Risk on meestel ja naistel sama.
Vanematel, kes on lähisugulased (verepilastus), on haigestunud lapse saamise võimalus suurem kui mitteseotud vanematel.
Domineerivad geneetilised häired tekivad siis, kui konkreetse haiguse tekitamiseks on vaja ainult ühte ebanormaalse geeni koopiat. Ebanormaalne geen võib pärida mõlemalt vanemalt või see võib olla mõjutatud isiku uue mutatsiooni (geenimuutuse) tulemus. Ebanormaalse geeni ülekandumise oht haigestunud vanemalt järglastele on 50% iga raseduse kohta. Risk on meestel ja naistel sama.
Mõnel inimesel on selle häire põhjuseks spontaanne geneetiline mutatsioon, mis esineb munas või spermas. Sellistes olukordades ei ole häire vanematelt päritud.
Mõjutatud populatsioonid
Autosomaalset retsessiivset sündroomi on kirjeldatud vähem kui 200 inimesel eri etnilistest rühmadest pärit peredes.
Autosomaalset domineerivat sündroomi on kirjeldatud vähem kui 50 peres.
Diagnostika
Robinoffi sündroomi diagnoos tehakse tavaliselt vahetult pärast sündi füüsiliste leidude, sealhulgas lühikese kasvu, jäsemete ja suguelundite anomaaliate ning näojoonte põhjal.
Geeni molekulaarne geneetiline testimine on saadaval autosomaalse retsessiivse Robinovi sündroomi diagnoosi kinnitamiseks ROR2. Geeni mutatsioonide molekulaarne geneetiline testimine WNT5A ja geen DVL1 saadaval autosomaalse domineeriva haiguse tüübi diagnoosi kinnitamiseks.
Robinovi sündroomi ravi
Robinovi sündroomi ravi keskendub konkreetsetele sümptomitele, mida iga inimene kogeb. Ravi võib nõuda spetsialistide meeskonna koordineeritud jõupingutusi. Lastearstid, skeletihäirete ravi spetsialistid (ortopeedid), kirurgid, südameprobleemide diagnoosimise ja ravi spetsialistid (kardioloogid), füsioterapeudid ja / või muud tervishoiuteenuse osutajad võivad vajada süstemaatilist ja terviklikku ravi planeerimist vigastatud laps.
Mõjutatud ebaselgete suguelunditega lastel saab soo vastsündinute perioodil kliinilise läbivaatuse abil õigesti määrata. Mõnel lapsel võidakse teha operatsioon ja / või võtta muid meetmeid krüptorhidismi ja / või muude suguelundite kõrvalekallete parandamiseks. Lisaks manustatakse mõnikord peenise pikkuse ja munandite mahu parandamiseks hormonaalseid toidulisandeid, nagu inimese kooriongonadotropiin ja / või testosteroon. Mõnel patsiendil on leitud kasvuhormooni puudulikkust, mis reageerib positiivselt kasvuhormoonravile. Neid hormoonravi vorme peab aga laste endokrinoloog hoolikalt jälgima.
Spetsiaalsed harjutused ja / või kirurgia võivad aidata teatud selgroohaiguste ravis. Operatsiooni võib teha ka kubemesongide, teatud kraniofasiaalsete kõrvalekallete, selgroo tõsise kõveruse (skolioos) korrigeerimiseks. seoses hemivertebra ja ribide, täielikult või osaliselt ühendatud sõrmede / varvaste (sündaktiliselt), huule- / suulaelõhe ja / või muude defektidega arengut. Traksid, hambakirurgia ja / või muud toetavad ravimeetodid võivad aidata parandada valesti asetatud hambaid ja / või muid suuõõne kõrvalekaldeid.
Loe ka:Düsleksia
Robinoffi sündroomiga imikud ja lapsed peavad viivitamatult tagama põhjaliku tervisekontrolli südamega seotud patoloogiate tuvastamine ja viivitamatu sobiv ravi, mis võivad sellega kaasneda häire. Mõjutatud imikuid ja lapsi tuleb samuti hoolikalt jälgida, et aidata kohe ära hoida ja / või avastada kopsuinfektsioone (kopsupõletik) ning pakkuda kiiret ja asjakohast ravi.
Robinovi sündroomiga laste varajane sekkumine on nende potentsiaali saavutamiseks hädavajalik. Eriteenused, mis võivad mõjutatud lastele abiks olla, võivad hõlmata parandusmeetmeid haridus, sotsiaalne tugi, füsioteraapia ja / või muu meditsiiniline, sotsiaalne ja / või professionaalsed teenused.
Prognoos
Robinovi sündroomi prognoos on üldiselt hea, kuid südamehaiguse raskusaste võib mõjutada eeldatavat eluiga.
Sarnased häired
Järgmiste häirete sümptomid võivad olla sarnased Robinovi sündroomiga. Võrdlused on diferentsiaaldiagnostika jaoks kasulikud:
AchondroplasiaKas pärilik haigus, mida iseloomustavad ebanormaalselt lühikesed käed ja jalad ning lühike kasv (lühikeste jäsemete kääbus), ebanormaalsed näojooned ja / või luustiku väärarengud. Näo tunnuste hulka võivad kuuluda ebanormaalselt suur pea (makrotsefaalia), otsmikupuu ebatavaline punnis, madal ninasild ja / või keskpinna alaareng (keskpinna hüpoplaasia).
Skeleti väärarengud võivad hõlmata ebatavaliselt lühikesi sõrmi ja varbaid (brachydactyly), ettepoole paisuvat selgroo sagitaalset painutust (lordoos), selgroo ahenemine (stenoos). Täiendavad häired võivad hõlmata probleeme küünarnuki ja puusa pikendamisega, lihastoonuse vähenemist (hüpotensioon) ja / või sagedasi keskkõrvapõletikke (keskkõrvapõletik).
Achondroplasia on autosoomne domineeriv geneetiline haigus.
Aarskogi sündroom on väga haruldane pärilik haigus, mida iseloomustavad pea ja näo kõrvalekalded (kraniofacial) piirkond, väike kuni mõõdukas lühike kasv, luustiku väärarengud ja / või suguelundite anomaaliad. Näo tunnuste hulka võivad kuuluda allapoole kallutatud silmad (silma hüpertelorism) silmalaugude voldid (pilud), väike nina koos ninasõõrmetega ja / või ülemise lõualuu vähearenenud areng (ülemise hüpoplaasia) lõualuu).
Skeleti väärarengud võivad hõlmata ebanormaalselt lühikesi varbaid (brachydactyly), viienda varba püsivat fikseerimist painutatud asendis (klinodactyly) ja / või ebatavaliselt laiad pöidlad. Suguelundite kõrvalekalded võivad hõlmata ebanormaalset nahavolt, mis ulatub ümber peenise aluse ja / või ühe või mõlema munandi võimetust laskuda munandikotti (krüptorhidism). Mõnedel lastel on kerge vaimne alaareng.
Aarskogi sündroom on X-seotud geneetiline haigus.
Omodüsplaasia See on haruldane pärilik luuhaigus, mida võib iseloomustada käte (nt õlavarreluu) mõnede pikkade luude (nt õlavarreluu) ja jalgade (nt reie) ebanormaalne lühis luud), lühikest kasvu ja iseloomulikke näojooni, sealhulgas allavajutatud ninasild, lai ninapõhi ja / või ebatavaliselt pikk vertikaalne soon (soon) ülaosa keskel huuled.
Lisaks võib kahjustatud lastel olla küünarvarre kahe luu ebanormaalne eraldumine (radiaalne diastaas), radiaalse pea nihestus ja / või ümara luumassi (kondüüli) alaareng (hüpoplaasia) lõpus õlg.
Omodüsplaasia võib pärida autosoomse retsessiivse või autosomaalse domineeriva geneetilise seisundina.


