Lipoidosis (lizoszómális lipidraktározási betegség): mi ez, példák
Figyelem! Az információ csak tájékoztató jellegű, és nem célja a diagnózis felállítása és a kezelés előírása. Mindig forduljon szakorvosához!
Tartalom
- Mik azok a lizoszómális raktározási betegségek?
- Mik azok a lipidek?
- Hogyan öröklődik a lipoidózis?
- A lizoszómális raktározási betegségek típusai és tünetei
Mik azok a lizoszómális raktározási betegségek?
Lizoszómális raktározási betegségek (lipoidózis), az örökletes anyagcsere-betegségek csoportja, amelyekben káros mennyiségű zsíros anyag (lipid) halmozódik fel a szervezet különböző sejtjeiben és szöveteiben.
Az ilyen betegségekben szenvedők vagy nem termelnek eleget a lipidek lebontásához (metabolizmusához) szükséges egyik enzimből, vagy olyan enzimeket termelnek, amelyek nem működnek megfelelően.
Idővel ez a túlzott zsírfelhalmozódás a sejtek és szövetek maradandó károsodásához vezethet, különösen az agyban, a perifériás idegrendszerben (az idegek a gerincvelőtől a többi részig test), máj, lép és a csontvelő.
Mik azok a lipidek?
A lipidek zsírszerű anyagok, amelyek fontos részei a sejteken belüli és sejtközi membránoknak, valamint az idegeket beborító és védő mielinhüvelynek. A lipidek közé tartoznak az olajok, zsírsavak, viaszok, szteroidok (például koleszterin és ösztrogén) és más rokon vegyületek.
Ezek a zsíros anyagok természetesen raktározódnak a szervezet sejtjeiben, szerveiben és szöveteiben. A sejten belüli apró testek, az úgynevezett lizoszómák, rendszeresen átalakítják vagy metabolizálják a lipideket és fehérjéket kisebb komponensekké, hogy energiát biztosítsanak a szervezetnek. Lizoszómális raktározási betegségeknek nevezzük azokat a rendellenességeket, amelyekben az intracelluláris, nem metabolizálható anyag a lizoszómákban marad.
A lipidek felhalmozódásával összefüggő betegségek mellett a felhalmozódással összefüggő egyéb lizoszómális betegségek közé tartoznak a mukolipidózisok, amelyekben a sejtek és szövetek felhalmozódnak. túlzott mennyiségű lipid, amelyhez cukormolekulák kapcsolódnak, valamint mukopoliszacharidózisok, amelyekben túlzott mennyiségű nagy, összetett cukor halmozódik fel molekulák.
Hogyan öröklődnek lipoidózis?
A lizoszómális raktározási betegségek az egyik vagy mindkét szülőtől öröklődnek, akik olyan hibás gént hordoznak, amely egy specifikus lipidmetabolizáló enzimet szabályoz a szervezet sejtjeinek egy osztályában. Kétféleképpen örökölhetők:
- Autoszomális recesszív öröklődés akkor fordul elő, ha mindkét szülő hordozza és továbbadja a hibás gén másolatát, de egyik szülőt sem érinti a rendellenesség. Minden gyermeknek, aki ezektől a szülőktől születik, 25 százalék az esélye, hogy örökli a hibás gén mindkét másolatát, 50 százalék annak a valószínűsége, hogy a szüleihez hasonló hordozó lesz, és 25 százalék az esélye annak, hogy nem örökli a hibás példány egyetlen példányát sem gén. Mindkét nemhez tartozó gyermekek autoszomális recesszív módon örökölhetők.
- X-hez kötött (vagy nemhez kapcsolódó) recesszív öröklődés akkor fordul elő, amikor az anya az érintett gént az X kromoszómán hordozza. Az X és Y kromoszómák részt vesznek a nemek meghatározásában. A nőknek két X kromoszómája van, míg a férfiaknak egy X kromoszómája és egy Y kromoszómája. A női hordozók fiai 50 százalékos eséllyel öröklik és befolyásolják a rendellenességet, mivel a fiúk egy X kromoszómát kapnak anyjuktól és egy Y kromoszómát az apjuktól. A lányok 50 százalékos eséllyel öröklik az érintett X-kromoszómát anyjuktól, és hordozók vagy enyhén érintettek. Az érintett férfiak nem adják át a betegséget fiaiknak, de lányaik hordozói és hordozói lesznek.
A lizoszómális raktározási betegségek típusai és tünetei
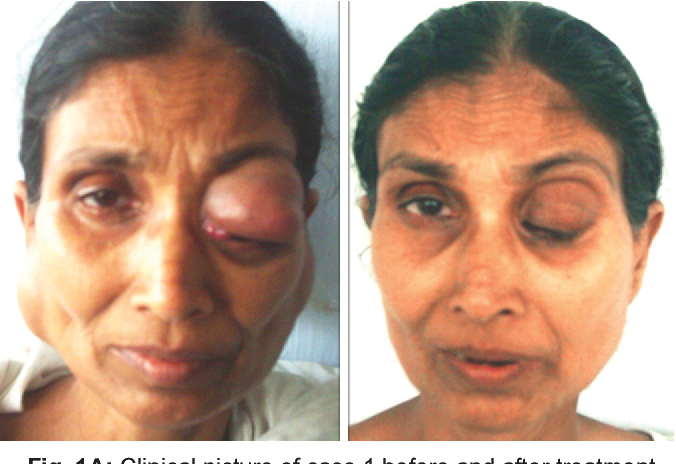
Gaucher-kór - a lizoszomális betegségek közül a leggyakoribb. A betegséget a glükocerebrozidáz enzim hiánya okozza, ami sok szövetben a glükocerebrozid felhalmozódásához vezet. A zsíros anyagok felhalmozódhatnak az agyban, a lépben, a májban, a vesékben, a tüdőben és a csontvelőben.
A tünetek közé tartozhat az agykárosodás, a lép megnagyobbodása és máj, károsodott májműködés, csontrendszeri rendellenességek és csontkárosodás, amely fájdalmat és töréseket, a nyirokcsomók és (néha) a szomszédos ízületek duzzadását okozhatja, puffadás, barnás bőrtónus, anémia, alacsony vérlemezkeszám és sárga foltok a szemen.
A legsúlyosabban érintett egyének érzékenyebbek lehetnek a fertőzésekre is. A betegség férfiakat és nőket egyaránt érint.
A Gaucher-kórnak három általános klinikai altípusa van:
- 1. típus (Vagy nem neuronopátiás típus) a betegség leggyakoribb formája az Egyesült Államokban és Európában. Az agyat nem érinti, de előfordulhat a tüdő és ritka esetekben a vese károsodása. A tünetek korai életkorban vagy felnőttkorban jelentkezhetnek, és magukban foglalhatják a máj megnagyobbodását és a lép súlyos megnagyobbodását, ami szakadáshoz és további szövődményekhez vezethet. A csontváz gyengesége és a csontbetegség kiterjedt lehet. Az ebbe a csoportba tartozó emberek általában könnyen zúzódások keletkeznek a vérük alacsony vérlemezkeszáma miatt. A vérszegénység miatt fáradtságot is tapasztalhatnak. A betegség kezdetétől és súlyosságától függően az emberek a típus 1 megélheti a felnőttkort. Sok szenvedőnek enyhe betegsége van, vagy előfordulhat, hogy nem mutatnak tüneteket. Bár 1típus A Gaucher-kór gyakori az askenázi zsidó örökséghez tartozó emberek körében, és bármilyen etnikai hátterű embert érinthet.
- 2. típus (vagy akut gyermekkori neuropátiás Gaucher-kór) általában a születést követő 3 hónapon belül kezdődik. A tünetek közé tartozik a kiterjedt és progresszív agykárosodás, görcsösség, görcsrohamok, merev végtagok, megnagyobbodott májmáj hepatomegalia) és lép (splenomegalia), kóros szemmozgás, valamint rossz szopási és nyelési képesség. Az érintett gyermekek általában 2 éves koruk előtt meghalnak.
- 3. típus (krónikus neuronopátiás forma) gyermekkorban vagy akár felnőttkorban bármikor elkezdődhet. Az akut Gaucher-kórhoz képest lassan progresszív, de kevésbé súlyos neurológiai tünetek jellemzik 2 féle. A fő tünetek közé tartoznak a szemmozgási zavarok, kognitív hiányosságok, rossz koordináció, görcsrohamok, a lép és/vagy a máj megnagyobbodása, csontrendszeri betegségek, vérbetegségek, beleértve a vérszegénységet és lélegző. Szinte mindenki Gaucher-kórban szenved 3 fajta, aki enzimpótló terápiában részesül, eléri a felnőttkort.
A betegek számára 1. és 3. típus A kéthetente intravénásan adott enzimpótló terápia drámaian csökkentheti a máj és a lép méretét, csökkentheti a csontrendszeri rendellenességeket és visszafordíthatja az egyéb megnyilvánulásokat. A sikeres csontvelő-transzplantáció kezeli a betegség neurológiai megnyilvánulásait. Ez az eljárás azonban jelentős kockázatokkal jár, és ritkán hajtják végre Gaucher-kórban szenvedő egyéneknél.
Ritka esetekben műtétre lehet szükség a lép egészének vagy egy részének eltávolítása érdekében (ha egy személynek nagyon alacsony a vérlemezkeszáma, vagy ha egy megnagyobbodott szerv nagymértékben befolyásolja a személy kényelmét). A vérátömlesztés néhány vérszegénységben szenvedő ember számára előnyös lehet. Másoknak ízületi helyettesítő műtétre lehet szükségük a mobilitás és az életminőség javítása érdekében. Jelenleg nincs hatékony kezelés a 2-es és 3-as típusú Gaucher-kórban szenvedőknél előforduló agykárosodásra.
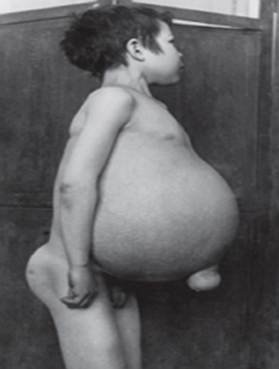
Niemann-Pick betegség Az autoszomális recesszív rendellenességek csoportja, amelyet a zsír és a koleszterin felhalmozódása okoz a máj, a lép, a csontvelő, a tüdő és bizonyos esetekben az agy sejtjeiben.
A neurológiai szövődmények közé tartozik az ataxia (az izomkoordináció hiánya, amely hatással lehet a járásra, írásra és étkezésre, egyéb funkciók mellett), szembénulás, agydegeneráció, tanulási problémák, görcsösség, táplálkozási és nyelési nehézség, elmosódott beszéd, izomtónus elvesztése, tapintásra való túlérzékenység és a szaruhártya bizonyos mértékű homályossága a túlzott felhalmozódás miatt anyagokat. Az érintettek 50%-ánál a retina közepe táján jellegzetes cseresznyevörös glória alakul ki, amelyet az orvos speciális műszerrel láthat.
A Niemann-Pick-kór három általános altípusra osztható:
- A típus, a legsúlyosabb forma, korai gyermekkorban kezdődik. A csecsemők születéskor normálisnak tűnnek, de 6 hónapos korukra mély agykárosodás alakul ki, megnagyobbodik a máj és a lép, valamint megduzzadnak a nyirokcsomók és a bőr alatti csomók (xantómák). A lép a normál méretének tízszeresére nőhet, és felszakadhat, vérzést okozva. Ezek a gyerekek fokozatosan gyengébbekké válnak, elveszítik a motoros funkciót, vérszegények lehetnek, és hajlamosak a visszatérő fertőzésekre. Ritkán élnek 18 hónapnál tovább. A betegség ezen formája leginkább a zsidó családokban fordul elő.
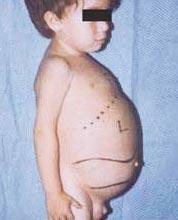
- B típus(vagy fiatalkori kezdet) általában nem érinti az agyat, de a legtöbb gyermek fejlődik ataxia, a gerincvelőt elhagyó idegek károsodása (perifériás neuropátia), valamint az életkorral előrehaladó tüdőproblémák. A máj és a lép megnagyobbodása jellemző a serdülőkre. Emberek B típus viszonylag sokáig élhetnek, de sokan egész életen át tartó oxigénterápiát igényelnek tüdőkárosodás miatt. A Niemann-Pick A és B típusa a szfingomielin nevű zsíranyag felhalmozódásának eredménye a szfingomielináz nevű enzim hiánya miatt.
- C típusú megjelenhet az élet korai szakaszában, vagy serdülőkorban vagy akár felnőttkorban is kialakulhat. Niemann-Pick betegségC típusú nem a szflingomielináz hiánya okozza, hanem az NPC1 vagy NPC2 fehérjék hiánya. Ennek eredményeként a különböző lipidek és különösen a koleszterin felhalmozódik az idegsejtekben, és működési zavarokat okoz. Az agy érintettsége kiterjedt lehet, ami képtelen fel-le nézni, járási és nyelési nehézségeket, progresszív hallásvesztést és progresszív hallásvesztést okozhat. elmebaj. Emberek C típusú csak enyhe lép és máj megnagyobbodása van. Az emberek várható élettartama C típusú jelentősen változik. Vannak, akik gyermekkorukban meghalnak, míg mások, akiket kevésbé érint, felnőttkorban is megélhetnek.
Jelenleg nincs gyógymód a Niemann-Pick-kórra. A kezelés támogató. A gyermekek általában fertőzés vagy progresszív neurológiai veszteség következtében halnak meg. Számos olyan betegnél fordultak elő csontvelő-transzplantáció B típus vegyes eredménnyel.
Fabry-kórmás néven alfa-galaktozidáz-A hiány, zsíros anyagok felhalmozódását okozza az autonóm idegrendszerben (az idegrendszer azon részében, szabályozza az önkéntelen funkciókat, mint a légzés és a szívverés), a szemben, a vesékben és a szív- és érrendszerben rendszer.
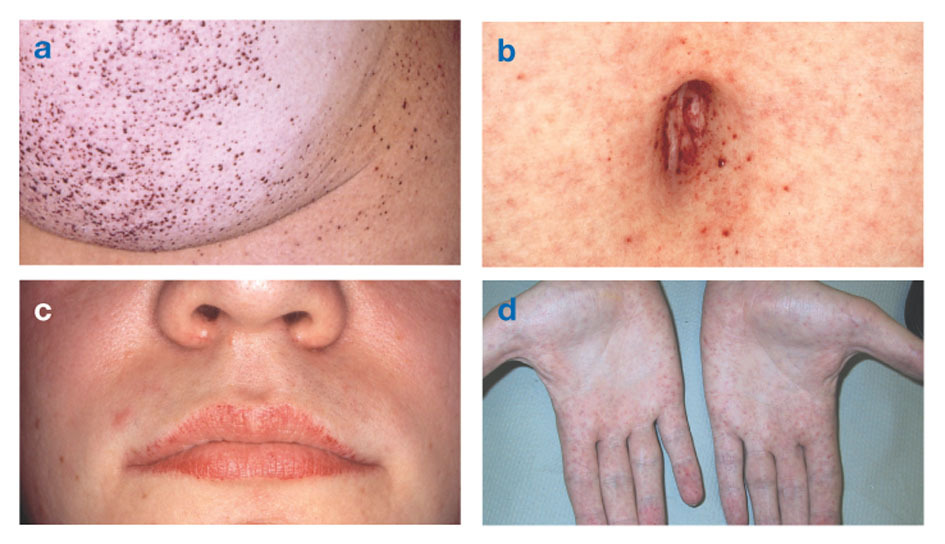
A Fabry-kór az egyetlen X-hez kötött lipidbetegség. A betegség elsősorban a férfiakat érinti, bár a nőknél enyhébb és változatosabb formája fordul elő. Ritkán az érintett nőknél olyan súlyos tünetek jelentkeznek, mint a betegségben szenvedő férfiaknál. A tünetek általában gyermekkorban vagy serdülőkorban jelentkeznek.
A neurológiai tünetek közé tartozik a karokban és lábakban fellépő égető fájdalom, amely meleg időben vagy azt követően fokozódik testmozgás, valamint a felesleges anyag felhalmozódása a szaruhártya átlátszó rétegeiben (ami homályosodáshoz vezet, de anélkül látásváltozások). Az erek falában felhalmozódó zsír megzavarhatja a vérkeringést, veszélybe sodorva az embert stroke vagy szívroham. Egyéb tünetek közé tartozik a szív megnagyobbodása, progresszív veseelégtelenségvégstádiumú vesebetegséghez, gyomor-bélrendszeri problémákhoz, csökkent izzadáshoz és lázhoz vezet.
Angiokeratómák (apró, nem rákos, vöröses-lilás domború foltok a bőrön) a törzs alsó részén alakulhatnak ki, és az életkorral egyre gyakoribbá válhatnak.
A Fabry-kórban szenvedők gyakran idő előtt meghalnak a betegség szövődményei miatt szív elégtelenségveseelégtelenség vagy szélütés. Az olyan gyógyszereket, mint a fenitoin és a karbamazepin, gyakran a Fabry-kórt kísérő fájdalom kezelésére írják fel, de nem a gyógyítására. A metoklopramid vagy a lipisorb (étrend-kiegészítő) enyhítheti a gyomor-bélrendszeri zavarokat gyakran fordul elő Fabry-kórban szenvedőknél, és néhány embernek veseátültetésre, ill dialízis. Az enzimpótlás csökkentheti a raktározást, enyhítheti a fájdalmat és megőrizheti a szervek működését egyes Fabry-kórban szenvedő betegeknél.
Farber-betegség más néven Farber-féle lipogranulomatosis, olyan ritka autoszomális recesszív rendellenességek csoportját írja le, amelyek zsíros anyagok felhalmozódását okozzák az ízületekben, szövetekben és a központi idegrendszerben. Férfiakat és nőket egyaránt érint. A betegség általában korai gyermekkorban kezdődik, de előfordulhat később is.
A klasszikus Farber-kórban szenvedő gyermekek kialakulnak neurológiai tünetek, amelyek magukban foglalhatják a fokozott álmosságot és levertséget, valamint problémákat nyelés. A máj, a szív és a vesék is érintettek lehetnek. Egyéb tünetek lehetnek ízületi kontraktúrák (az ízületek körüli izmok vagy inak krónikus megrövidülése), hányás, ízületi gyulladás, megnagyobbodás nyirokcsomók, ízületi gyulladás, rekedtség és bőr alatti csomók, amelyek előrehaladtával megvastagodnak az ízületek körül betegségek.
Légzési nehézségekkel küzdő betegeknél előfordulhat, hogy légzőcsövet kell behelyezni. A legtöbb ilyen betegségben szenvedő gyermek 2 éves korára meghal, általában az tüdőbetegségek. A betegség egyik legsúlyosabb formájában a születés után röviddel máj- és lépmegnagyobbodás diagnosztizálható. A betegség ezen formájával született babák általában 6 hónapon belül meghalnak.
A Farber-kórt a ceramidáz nevű enzim hiánya okozza. Jelenleg nincs specifikus kezelés a Farber-kórra. A fájdalom enyhítésére kortikoszteroidokat írhatnak fel. A csontvelő-transzplantáció csökkentheti a granulómákat (kis mennyiségű gyulladt szövet) enyhe gyulladásban szenvedő betegeknél, illetve olyan betegeknél, akiknél nem fordul elő tüdő- vagy idegrendszeri szövődmény. Időseknél a granulomák zsugoríthatók vagy műtéti úton eltávolíthatók.
Gangliozidózis genetikai betegségek két különböző csoportjából áll. Mindkettő autoszomális recesszív, és egyformán érinti a férfiakat és a nőket.
GM1 gangliozidózis.
A GM1 gangliozidózist a béta-galaktozidáz enzim hiánya okozza, ami abnormális felhalmozódást eredményez. savas lipid anyagok, különösen a központi és a perifériás idegrendszer idegsejtjeiben rendszerek. A GM1 gangliozidózisnak három klinikai megnyilvánulása van:
- GM1 (a legsúlyosabb altípus, amely röviddel a születés után jelentkezik) tartalmazhat neurodegenerációt, görcsrohamokat, máj- és lépmegnagyobbodást, az arcvonások durvasága, a csontváz egyenetlenségei, az ízületi merevség, a puffadás, az izomgyengeség, a túlzott riasztási reakció és a testtartás. Az érintettek körülbelül felénél cseresznyevörös foltok jelennek meg a szemben. A gyerekek 1 éves korukra süketek és vakok lehetnek, és gyakran meghalnak 3 éves korukra szívszövődmények vagy tüdőgyulladás.
- Késő gyerek A GM1 gangliozidózis általában 1 és 3 éves kor között kezdődik. A neurológiai tünetek közé tartozik az ataxia, görcsrohamok, demencia és beszédzavar.
- GM1 gangliozidózis 3 és 30 éves kor között alakul ki. A tünetek közé tartozik az izomtömeg csökkenése (izom atrófia), a neurológiai szövődmények, amelyek kevésbé súlyosak és lassabban fejlődnek, mint más formák rendellenességek, egyes embereknél a szaruhártya homályossága és tartós izomösszehúzódások, amelyek csavarodó és ismétlődő mozdulatokat vagy rendellenes testtartást okoznak (disztónia). Angiokeratómák alakulhatnak ki a test alsó részén. A máj és a lép mérete a legtöbb érintett embernél normális.
GM2 gangliozidózis.
A GM2 gangliozidózis azt is okozza, hogy a szervezet felesleges savas zsírokat halmoz fel a szövetekben és sejtekben, elsősorban az idegsejtekben. Ezek a rendellenességek a béta-hexózaminidáz enzim hiányából erednek. A GM2 rendellenességek közé tartoznak:
- Tay-Sachs betegség (más néven GM2 gangliozidózis B változat) és variáns formáit a hexosaminidáz A enzim hiánya okozza. Az érintett gyermekek normálisan fejlődnek életük első hónapjaiban. A tünetek 6 hónapos korban kezdődnek, és magukban foglalják a mentális képességek fokozatos elvesztését, demencia, csökkent szemkontaktus, fokozott megdöbbentő reakció a zajra, süketséghez vezető progresszív hallásvesztés, nyelési nehézség, vakság, cseresznyevörös foltok a retinán és néhány bénulás. A rohamok a gyermek életének második évében kezdődhetnek. A csecsemők végül függővé válhatnak az etetőcsőtől, és gyakran 4 éves korukra belehalnak a visszatérő fertőzésekbe. Sajnos nincs speciális kezelés. A görcsoldók kezdetben kontrollálhatják a rohamokat. Egyéb támogató kezelések közé tartozik a megfelelő táplálkozás és hidratálás, valamint a légutak nyitva tartásának módszerei. A rendellenesség ritka formája az ún későn jelentkező Tay-Sachs-kór, 20 és 30 év közötti embereknél fordul elő, instabil járás és progresszív neurológiai leromlás jellemzi.
- Sandhoff-kór (AB lehetőség) a Tay-Sachs-betegség súlyos formája. Általában 6 hónapos korban jelentkezik, és nem korlátozódik egyetlen etnikai csoportra sem. A neurológiai tünetek közé tartozhat a központi idegrendszer, a motoros állapot progresszív romlása gyengeség, korai vakság, súlyos ijedt reakció hangra, görcsösség, sokk vagy rángatózás izmok (myoclonus), epilepszia, kórosan megnagyobbodott fej (makrokefália) és vörös foltok a szemben. Egyéb tünetek közé tartozhatnak a gyakori légúti fertőzések, szívzörej, babaszerű arcvonások és a máj és a lép megnagyobbodása. A Sandhoff-kórra nincs specifikus kezelés. A Tay-Sachs-kórhoz hasonlóan a szupportív ellátás magában foglalja a légutak állandó nyitva tartását, valamint a megfelelő táplálkozást és hidratálást. Az antikonvulzív szerek kezdetben az epilepsziát (görcsrohamokat) tudják kontrollálni.
Krabbe-betegség(más néven globuláris sejt leukodystrophia és galaktozilceramid lipidózis) egy autoszomális recesszív betegség, amelyet a galaktocerebrozidáz enzim hiánya okoz. A betegség leggyakrabban a 6 hónapos kor előtti csecsemőket érinti, de előfordulhat serdülőkorban vagy felnőttkorban is.
Az emésztetlen zsírok felhalmozódása befolyásolja az ideg védőburkolatának (mielinhüvely) növekedését, és súlyos mentális és motoros károsodást okoz. Egyéb tünetek közé tartozik az izomgyengeség, az izmok csökkent nyúlási képessége (hipotónia), izomfeszülés (spaszticitás), hirtelen sokk vagy a végtagok rángatózása (mioklónusos rohamok), ingerlékenység, megmagyarázhatatlan láz, süketség, vakság, bénulás és nyelési nehézség. Hosszú távú fogyás is előfordulhat.
A betegség enzimvizsgálattal és jellegzetes sejtcsoportjának azonosításával diagnosztizálható globoid testekké az agy fehérállományában, az idegek demielinizációja és degenerációja, valamint az agysejtek pusztulása. Csecsemőknél a betegség általában 2 éves koruk előtt végzetes. A betegség előrehaladottabb formájában szenvedők enyhébb lefolyásúak, és jelentősen tovább élnek.
A Krabbe-kór specifikus kezelését nem fejlesztették ki, bár a korai csontvelő-transzplantáció segíthet néhány embernek. A betegség előrehaladottabb formájában szenvedők enyhébb lefolyásúak, és jelentősen tovább élnek.
Metakromatikus leukodystrophiaAz MLD a központi idegrendszer fehérállományában, a perifériás idegekben és bizonyos mértékig a vesékben felhalmozódó zsíros anyagokkal jellemezhető rendellenességek csoportja. A Krabbe-kórhoz hasonlóan az MLD a mielint érinti, amely lefedi és védi az idegeket. Ezt az autoszomális recesszív rendellenességet az arilszulfatáz A enzim hiánya okozza. Ez a rendellenesség férfiakat és nőket egyaránt érint.
A metakromatikus leukodystrophiának három jellegzetes formája van: késői csecsemő, fiatalkori és felnőtt.
- Késői csecsemő MLD általában a születés után 12-20 hónappal kezdődik. A gyermekek eleinte normálisnak tűnhetnek, de nehezen járnak, és hajlamosak elesni, amit időszakos fájdalom kísér a karokban és lábakban. progresszív látásvesztés, ami vaksághoz, fejlődési késésekhez és a korábban megszerzett mérföldkövek elvesztéséhez, nyelési zavarokhoz, görcsrohamokhoz és demenciához vezet 2-ig évek. A gyermekekben is progresszív izomsorvadás és gyengeség alakul ki, és végül elveszítik járási képességüket. A legtöbb ilyen betegségben szenvedő gyermek 5 éves korára meghal.
- Fiatalkorú MLD általában 3 és 10 éves kor között kezdődik. A tünetek közé tartozik az iskolai teljesítmény romlása, a mentális károsodás, az ataxia, a görcsrohamok és a demencia. A tünetek előrehaladnak, a halálozás 10-20 évvel a betegség kezdete után következik be.
- Felnőtt forma. Tünetek felnőttek 16 éves kor után kezdődik, és magában foglalhat ataxiát, görcsrohamokat, kóros végtagremegést (remegés), koncentrációs zavarokat, depresszió, mentális zavarok és demencia. A halál általában 6-14 évvel a tünetek megjelenése után következik be.
Az MLD nem gyógyítható. A kezelés tüneti és támogató. A csontvelő-transzplantáció bizonyos esetekben késleltetheti a betegség előrehaladását. Jelentős előrelépés történt a génterápia terén MLD-vel rendelkező állatmodellek és klinikai vizsgálatok során.
Wolman-kórmás néven lizoszómális savas lipáz hiánysúlyos lipidraktározási zavar, amely általában 1 éves korban végzetes. Ezt az autoszomális recesszív rendellenességet a koleszterin-észterek (általában a koleszterin transzport formája) felhalmozódása jellemzi. trigliceridek (az a kémiai forma, amelyben a zsírok léteznek a szervezetben), amelyek jelentősen felhalmozódhatnak és sejtkárosodást okozhatnak és szövetek.
Mind a férfiak, mind a nők szenvednek ettől a rendellenességtől. A csecsemők születésükkor normálisak és aktívak, de gyorsan fejlődnek náluk progresszív mentális károsodás, megnagyobbodott máj és nagymértékben megnagyobbodott lép, puffadás, gyomor-bélrendszeri problémák. sárgaság, vérszegénység, hányás és kalcium lerakódások mellékvesékmegkeményedését okozva.
Egy másik típus lizoszómasav lipáz hiánya az koleszterin-észter tárolási betegség. Ez a rendkívül ritka betegség a koleszterin észterek raktározása és trigliceridek a vérsejtekben, nyirok és nyirokszövet. Gyermekeknél felnőttkorban a máj megnagyobbodik, ami cirrhosishoz és krónikus májelégtelenség. A gyermekeknél a mellékvesékben is előfordulhatnak kalciumlerakódások, a betegség későbbi szakaszaiban sárgaság is kialakulhat.
Jelenleg az enzimpótlás kérdését mind a Wolman-kórban, mind a koleszterin-észter felhalmozódásában aktívan tanulmányozzák.
Figyelem! Az információ csak tájékoztató jellegű, és nem célja a diagnózis felállítása és a kezelés előírása. Mindig forduljon szakorvosához!