Lipoidosis (Penyakit penyimpanan lipid lisosom): apa itu, contoh
Perhatian! Informasi ini hanya untuk tujuan informasi dan tidak dimaksudkan untuk mendiagnosis dan meresepkan pengobatan. Selalu konsultasikan dengan dokter spesialis Anda!
Isi
- Apa itu penyakit penyimpanan lisosom?
- Apa itu lipid?
- Bagaimana lipoidosis diturunkan?
- Jenis dan gejala penyakit penyimpanan lisosom
Apa itu penyakit penyimpanan lisosom?
Penyakit penyimpanan lisosom (lipoidosis), adalah sekelompok penyakit metabolik herediter di mana sejumlah besar zat lemak (lipid) yang berbahaya menumpuk di berbagai sel dan jaringan tubuh.
Orang dengan kelainan ini tidak membuat cukup salah satu enzim yang dibutuhkan untuk memecah (memetabolisme) lipid, atau mereka menghasilkan enzim yang tidak bekerja dengan baik.
Seiring waktu, akumulasi lemak yang berlebihan ini dapat menyebabkan kerusakan permanen pada sel dan jaringan, terutama di otak, sistem saraf tepi (saraf dari sumsum tulang belakang ke seluruh) tubuh), hati, limpa dan sumsum tulang.
Apa itu lipid?
Lipid adalah zat seperti lemak yang merupakan bagian penting dari membran di dalam dan di antara sel dan dalam selubung mielin, yang menutupi dan melindungi saraf. Lipid termasuk minyak, asam lemak, lilin, steroid (seperti kolesterol dan estrogen), dan senyawa terkait lainnya.
Zat lemak ini secara alami disimpan di dalam sel, organ dan jaringan tubuh. Tubuh kecil di dalam sel, yang disebut lisosom, secara teratur mengubah atau memetabolisme lipid dan protein menjadi komponen yang lebih kecil untuk menyediakan energi bagi tubuh. Gangguan di mana bahan intraseluler, yang tidak dapat dimetabolisme, tetap berada di lisosom, disebut penyakit penyimpanan lisosom.
Selain penyakit yang terkait dengan akumulasi lipid, penyakit lisosom lain yang terkait dengan akumulasi termasuk mukolipidosis, di mana sel dan jaringan menumpuk. jumlah lipid yang berlebihan dengan molekul gula yang melekat padanya, serta mucopolysaccharidosis, di mana sejumlah besar gula kompleks terakumulasi molekul.
Bagaimana diwariskan? lipoidosis?
Penyakit penyimpanan lisosom diturunkan dari salah satu atau kedua orang tua yang membawa gen cacat yang mengatur enzim metabolisme lipid tertentu dalam kelas sel dalam tubuh. Mereka dapat diwarisi dengan dua cara:
- Warisan resesif autosomal terjadi ketika kedua orang tua membawa dan meneruskan salinan gen yang rusak, tetapi tidak ada orang tua yang terpengaruh oleh kelainan tersebut. Setiap anak yang lahir dari orang tua ini memiliki peluang 25 persen untuk mewarisi kedua salinan gen yang rusak, 50 persen kemungkinan bahwa ia akan menjadi pembawa yang mirip dengan orang tuanya, dan kemungkinan 25 persen untuk tidak mewarisi salinan apa pun dari yang cacat gen. Anak-anak dari kedua jenis kelamin dapat diwariskan secara resesif autosomal.
- Warisan resesif terkait-X (atau terkait seks) terjadi ketika ibu membawa gen yang terkena pada kromosom X. Kromosom X dan Y terlibat dalam penentuan jenis kelamin. Wanita memiliki dua kromosom X, sedangkan pria memiliki satu kromosom X dan satu kromosom Y. Anak laki-laki dari pembawa perempuan memiliki peluang 50 persen untuk mewarisi dan mempengaruhi kelainan tersebut, karena anak laki-laki menerima satu kromosom X dari ibu mereka dan satu kromosom Y dari ayah mereka. Anak perempuan memiliki peluang 50 persen untuk mewarisi kromosom X yang terpengaruh dari ibu mereka dan merupakan pembawa atau sedikit terpengaruh. Laki-laki yang terkena tidak menularkan penyakit kepada anak laki-laki mereka, tetapi anak perempuan mereka akan menjadi pembawa dan pembawa penyakit.
Jenis dan gejala penyakit penyimpanan lisosom
penyakit Gaucher - penyakit lisosom yang paling umum. Penyakit ini disebabkan oleh defisiensi enzim glukoserebrosidase, yang menyebabkan akumulasi glukoserebrosida di banyak jaringan. Bahan lemak dapat menumpuk di otak, limpa, hati, ginjal, paru-paru, dan sumsum tulang.
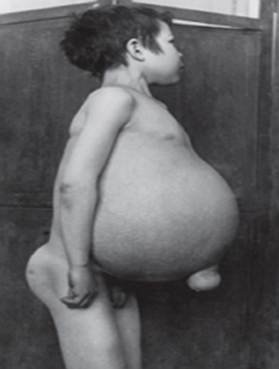
Gejala mungkin termasuk kerusakan otak, pembesaran limpa dan hati, gangguan fungsi hati, gangguan tulang dan kerusakan tulang yang dapat menyebabkan nyeri dan patah tulang, pembengkakan kelenjar getah bening dan (kadang-kadang) sendi yang berdekatan, kembung, warna kulit kecoklatan, anemia, jumlah trombosit yang rendah dan bintik-bintik kuning pada mata.
Individu yang paling parah terkena dampak mungkin juga lebih rentan terhadap infeksi. Penyakit ini mempengaruhi pria dan wanita secara setara.
Penyakit Gaucher memiliki tiga subtipe klinis umum:
- Tipe 1 (Atau tidak neuropatik type) adalah bentuk penyakit yang paling umum di Amerika Serikat dan Eropa. Otak tidak terpengaruh, tetapi mungkin ada kerusakan pada paru-paru dan, dalam kasus yang jarang terjadi, ginjal. Gejala dapat dimulai pada awal kehidupan atau hingga dewasa dan termasuk pembesaran hati dan pembesaran limpa yang parah, yang dapat menyebabkan ruptur dan komplikasi tambahan. Kelemahan rangka dan penyakit tulang bisa meluas. Orang-orang dalam kelompok ini biasanya mudah memar karena jumlah trombosit yang rendah dalam darah mereka. Mereka mungkin juga mengalami kelelahan karena anemia. Tergantung pada onset dan keparahan penyakit, orang dengan tipe 1 dapat hidup sampai dewasa. Banyak penderita memiliki penyakit ringan atau mungkin tidak menunjukkan gejala apapun. Meskipun 1Tipe Penyakit Gaucher adalah umum di antara orang-orang dari keturunan Yahudi Ashkenazi dan dapat mempengaruhi orang-orang dari semua latar belakang etnis.
- Tipe 2 (atau neuropatik anak akut penyakit Gaucher) biasanya dimulai dalam waktu 3 bulan setelah lahir. Gejalanya meliputi kerusakan otak yang luas dan progresif, spastisitas, kejang, tungkai kaku, pembesaran hati (hepatomegali hati) dan limpa (splenomegali), gerakan mata yang tidak normal, dan kemampuan mengisap dan menelan yang buruk. Anak-anak yang terkena biasanya meninggal sebelum usia 2 tahun.
- Tipe 3 (Neuropatik kronis bentuk) dapat dimulai kapan saja selama masa kanak-kanak atau bahkan di masa dewasa. Hal ini ditandai dengan gejala neurologis yang progresif lambat tetapi kurang parah dibandingkan dengan penyakit Gaucher akut 2 jenis. Gejala utama termasuk gangguan gerakan mata, defisit kognitif, koordinasi yang buruk, kejang, pembesaran limpa dan/atau hati, kelainan tulang, kelainan darah termasuk anemia, dan masalah dengan pernafasan. Hampir semua orang dengan penyakit Gaucher 3 jenis, yang menerima terapi penggantian enzim akan mencapai usia dewasa.
Untuk pasien Tipe 1 dan 3 Terapi penggantian enzim, yang diberikan secara intravena setiap dua minggu, dapat secara dramatis mengurangi ukuran hati dan limpa, mengurangi kelainan tulang, dan membalikkan manifestasi lainnya. Transplantasi sumsum tulang yang berhasil mengobati manifestasi neurologis penyakit. Namun, prosedur ini membawa risiko yang signifikan dan jarang dilakukan pada individu dengan penyakit Gaucher.
Dalam kasus yang jarang terjadi, pembedahan mungkin diperlukan untuk mengangkat seluruh atau sebagian limpa (jika seseorang memiliki jumlah trombosit yang sangat rendah atau ketika organ yang membesar sangat mempengaruhi kenyamanan orang tersebut). Transfusi darah mungkin bermanfaat bagi beberapa orang dengan anemia. Orang lain mungkin memerlukan operasi penggantian sendi untuk meningkatkan mobilitas dan kualitas hidup. Saat ini tidak ada pengobatan yang efektif untuk kerusakan otak yang dapat terjadi pada orang dengan penyakit Gaucher tipe 2 dan 3.
Penyakit Niemann-Pick adalah sekelompok gangguan resesif autosomal yang disebabkan oleh akumulasi lemak dan kolesterol dalam sel-sel hati, limpa, sumsum tulang, paru-paru dan, dalam beberapa kasus, otak.
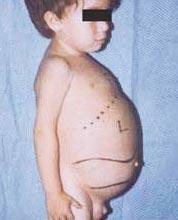
Komplikasi neurologis dapat mencakup ataksia (kurangnya koordinasi otot yang dapat mempengaruhi berjalan, menulis dan makan, antara lain), kelumpuhan mata, degenerasi otak, masalah belajar, kelenturan, kesulitan makan dan menelan, cadel bicara, hilangnya tonus otot, hipersensitivitas terhadap sentuhan, dan beberapa kekeruhan kornea karena akumulasi berlebihan bahan. Pada 50% orang yang terkena, lingkaran merah ceri yang khas berkembang di sekitar pusat retina, yang dapat dilihat oleh dokter dengan alat khusus.
Penyakit Niemann-Pick diklasifikasikan menjadi tiga subtipe umum:
- Tipe A, bentuk yang paling parah, dimulai pada anak usia dini. Bayi tampak normal saat lahir, tetapi pada usia 6 bulan, terjadi kerusakan otak dalam, pembesaran hati dan limpa, serta pembengkakan kelenjar getah bening dan kelenjar di bawah kulit (xantoma). Limpa bisa menjadi 10 kali ukuran normalnya dan bisa pecah, menyebabkan pendarahan. Anak-anak ini menjadi semakin lemah, kehilangan fungsi motorik, dapat menjadi anemia, dan rentan terhadap infeksi berulang. Mereka jarang hidup lebih dari 18 bulan. Bentuk penyakit ini paling sering terjadi pada keluarga Yahudi.
- Tipe B(atau onset remaja) biasanya tidak mempengaruhi otak, tetapi kebanyakan anak-anak berkembang ataxia, kerusakan pada saraf yang meninggalkan sumsum tulang belakang (neuropati perifer), dan kesulitan paru yang berkembang seiring bertambahnya usia. Pembesaran hati dan limpa khas untuk remaja. Orang dengan tipe B dapat hidup relatif lama, tetapi banyak yang membutuhkan terapi oksigen seumur hidup karena kerusakan paru-paru. Niemann-Pick tipe A dan B adalah hasil akumulasi dari zat lemak yang disebut sphingomyelin karena kekurangan enzim yang disebut sphingomyelinase.
- Tipe C mungkin muncul di awal kehidupan atau berkembang selama masa remaja atau bahkan dewasa. Penyakit Niemann-Picktipe C disebabkan bukan oleh defisiensi sphlingomyelinase, tetapi oleh kekurangan protein NPC1 atau NPC2. Akibatnya, berbagai lipid dan terutama kolesterol menumpuk di sel saraf dan menyebabkan kerusakannya. Keterlibatan otak bisa luas, mengakibatkan ketidakmampuan untuk melihat ke atas dan ke bawah, kesulitan berjalan dan menelan, gangguan pendengaran progresif, dan gangguan pendengaran progresif. demensia. Orang dengan tipe C hanya mengalami pembesaran limpa dan hati yang ringan. Harapan hidup orang dengan tipe C sangat bervariasi. Beberapa orang meninggal di masa kanak-kanak, sementara yang lain yang tidak terlalu terpengaruh mungkin juga hidup di masa dewasa.
Saat ini tidak ada obat untuk penyakit Niemann-Pick. Pengobatan bersifat suportif. Anak-anak biasanya meninggal karena infeksi atau kehilangan neurologis progresif. Ada kasus transplantasi sumsum tulang pada beberapa pasien dengan tipe B dengan hasil yang beragam.
Penyakit Fabryjuga dikenal sebagai defisiensi alfa-galaktosidase-A, menyebabkan akumulasi materi lemak dalam sistem saraf otonom (bagian dari sistem saraf yang mengontrol fungsi tak sadar seperti pernapasan dan detak jantung), di mata, ginjal, dan kardiovaskular sistem.
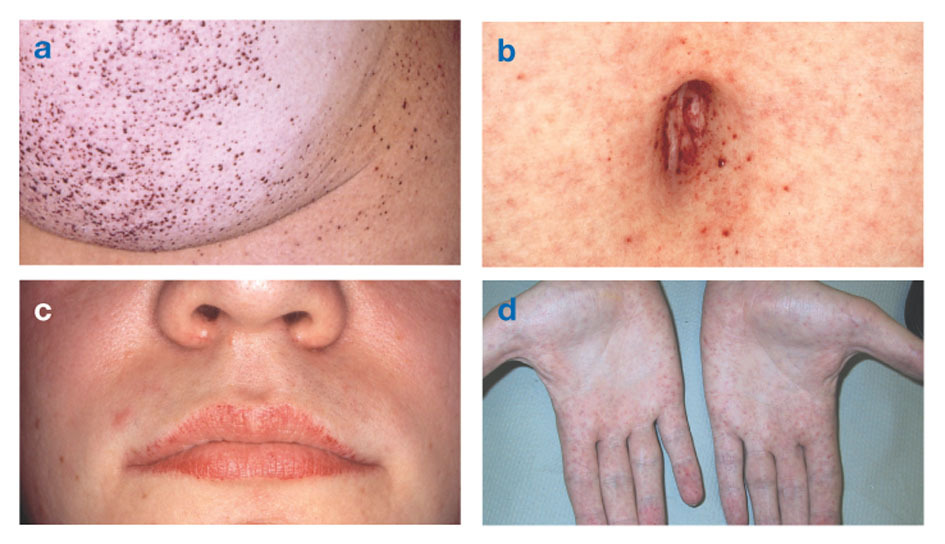
Penyakit Fabry adalah satu-satunya penyakit lipid terkait-X. Penyakit ini terutama menyerang pria, meskipun bentuk yang lebih ringan dan lebih bervariasi terjadi pada wanita. Jarang, wanita yang terkena memiliki gejala parah yang mirip dengan yang terlihat pada pria dengan gangguan tersebut. Timbulnya gejala biasanya terjadi selama masa kanak-kanak atau remaja.
Tanda-tanda neurologis termasuk rasa sakit yang membakar di lengan dan kaki yang memburuk saat cuaca panas atau setelahnya olahraga, serta akumulasi bahan berlebih di lapisan transparan kornea (yang menyebabkan kekeruhan, tetapi tanpa perubahan penglihatan). Penumpukan lemak di dinding pembuluh darah dapat mengganggu sirkulasi, menempatkan seseorang pada risiko pukulan atau serangan jantung. Gejala lain termasuk pembesaran jantung, progresif gagal ginjalmenyebabkan penyakit ginjal stadium akhir, masalah pencernaan, penurunan keringat dan demam.
Angiokeratoma (tambalan kecil, non-kanker, berwarna ungu kemerahan pada kulit) dapat berkembang di tubuh bagian bawah dan menjadi lebih umum seiring bertambahnya usia.
Orang dengan penyakit Fabry sering meninggal sebelum waktunya akibat komplikasi dari: gagal jantung, gagal ginjal, atau stroke. Obat-obatan seperti fenitoin dan karbamazepin sering diresepkan untuk mengobati rasa sakit yang menyertai penyakit Fabry, tetapi tidak untuk menyembuhkannya. Metoclopramide atau Lipisorb (suplemen makanan) dapat meredakan gangguan gastrointestinal yang sering terjadi pada orang dengan penyakit Fabry, dan beberapa orang mungkin memerlukan transplantasi ginjal atau dialisis. Penggantian enzim dapat mengurangi penyimpanan, menghilangkan rasa sakit, dan mempertahankan fungsi organ pada beberapa orang dengan penyakit Fabry.
penyakit farber juga dikenal sebagai lipogranulomatosis Farber, menggambarkan sekelompok gangguan resesif autosomal langka yang menyebabkan akumulasi materi lemak di sendi, jaringan, dan sistem saraf pusat. Ini mempengaruhi pria dan wanita. Penyakit ini biasanya dimulai pada anak usia dini, tetapi dapat terjadi di kemudian hari.
Anak-anak dengan penyakit Farber klasik berkembang gejala neurologis, yang mungkin termasuk peningkatan kantuk dan kelesuan, dan masalah dengan menelan. Hati, jantung, dan ginjal juga dapat terpengaruh. Gejala lain mungkin termasuk kontraktur sendi (pemendekan kronis otot atau tendon di sekitar sendi), muntah, radang sendi, pembesaran kelenjar getah bening, radang sendi, suara serak, dan benjolan di bawah kulit yang menebal di sekitar sendi saat berkembang penyakit.
Orang yang terkena dengan kesulitan bernapas mungkin perlu memasukkan tabung pernapasan. Sebagian besar anak dengan kondisi ini meninggal pada usia 2 tahun, biasanya karena penyakit paru paru. Dalam salah satu bentuk penyakit yang paling parah, pembesaran hati dan limpa dapat didiagnosis segera setelah lahir. Bayi yang lahir dengan bentuk penyakit ini biasanya meninggal dalam waktu 6 bulan.
Penyakit Farber disebabkan oleh kekurangan enzim yang disebut ceramidase. Saat ini tidak ada pengobatan khusus untuk penyakit Farber. Kortikosteroid dapat diresepkan untuk menghilangkan rasa sakit. Transplantasi sumsum tulang dapat mengurangi granuloma (massa kecil jaringan yang meradang) pada orang dengan peradangan ringan atau pada pasien tanpa komplikasi paru atau sistem saraf. Pada orang tua, granuloma dapat menyusut atau diangkat melalui pembedahan.
Gangliosidosis terdiri dari dua kelompok penyakit genetik yang berbeda. Keduanya resesif autosomal dan mempengaruhi pria dan wanita secara setara.
gangliosidosis GM1.
Gangliosidosis GM1 disebabkan oleh defisiensi enzim beta-galaktosidase, yang mengakibatkan akumulasi abnormal dari bahan lipid asam, khususnya pada sel-sel saraf di saraf pusat dan perifer sistem. Gangliosidosis GM1 memiliki tiga manifestasi klinis:
- GM1 (subtipe yang paling parah, terjadi segera setelah lahir) mungkin termasuk neurodegenerasi, kejang, pembesaran hati dan limpa, kekasaran fitur wajah, ketidakteraturan tulang, kekakuan sendi, kembung, kelemahan otot, respons kaget yang berlebihan, dan masalah dengan kiprah. Sekitar setengah dari mereka yang terkena mengembangkan bintik-bintik merah ceri di mata. Anak-anak mungkin tuli dan buta pada usia 1 tahun dan sering meninggal pada usia 3 tahun karena komplikasi jantung atau radang paru-paru.
- anak terlambat Gangliosidosis GM1 biasanya dimulai antara usia 1 dan 3 tahun. Gejala neurologis termasuk ataksia, kejang, demensia, dan kesulitan berbicara.
- Gangliosidosis GM1 berkembang antara usia 3 dan 30 tahun. Gejalanya meliputi penurunan massa otot (atrofi otot), komplikasi neurologis yang tidak terlalu parah dan berkembang lebih lambat daripada bentuk lainnya gangguan, kekeruhan kornea pada beberapa orang, dan kontraksi otot berkelanjutan yang menyebabkan gerakan memutar dan berulang atau postur abnormal (distonia). Angiokeratoma dapat berkembang di batang tubuh bagian bawah. Ukuran hati dan limpa normal pada kebanyakan orang yang terkena.
gangliosidosis GM2.
Gangliosidosis GM2 juga menyebabkan tubuh menumpuk zat lemak asam berlebih di jaringan dan sel, terutama sel saraf. Gangguan ini hasil dari kekurangan enzim beta-hexosaminidase. Gangguan GM2 meliputi:
- penyakit Tay Sachs (juga dikenal sebagai GM2 gangliosidosis varian B) dan bentuk variannya disebabkan oleh defisiensi enzim hexosaminidase A. Anak-anak yang terkena berkembang secara normal selama beberapa bulan pertama kehidupan. Gejala dimulai pada usia 6 bulan dan termasuk hilangnya kapasitas mental secara progresif, demensia, penurunan kontak mata, peningkatan respons mengejutkan terhadap kebisingan, gangguan pendengaran progresif yang menyebabkan tuli, kesulitan menelan, kebutaan, bintik merah ceri pada retina, dan beberapa kelumpuhan. Kejang dapat dimulai pada tahun kedua kehidupan seorang anak. Bayi pada akhirnya dapat menjadi tergantung pada selang makanan, dan sering meninggal pada usia 4 tahun karena infeksi berulang. Sayangnya, tidak ada perlakuan khusus. Antikonvulsan awalnya dapat mengontrol kejang. Perawatan suportif lainnya termasuk nutrisi dan hidrasi yang tepat, dan metode untuk menjaga saluran udara tetap terbuka. Suatu bentuk kelainan langka yang disebut penyakit Tay-Sachs onset lambat, terjadi pada orang antara usia 20 dan 30 dan ditandai dengan gaya berjalan yang tidak stabil dan kerusakan neurologis progresif.
- penyakit Sandhoff (Opsi AB) adalah bentuk parah dari penyakit Tay-Sachs. Onset biasanya terjadi pada usia 6 bulan dan tidak terbatas pada kelompok etnis manapun. Tanda-tanda neurologis mungkin termasuk kerusakan progresif sistem saraf pusat, gangguan motorik, kelemahan, kebutaan dini, reaksi ketakutan yang parah terhadap suara, kelenturan, syok, atau kedutan otot (mioklonus), epilepsi, kepala yang membesar secara tidak normal (macrocephaly), dan bercak merah di mata. Gejala lain mungkin termasuk infeksi pernapasan yang sering, bisikan hati, fitur wajah seperti boneka dan pembesaran hati dan limpa. Tidak ada pengobatan khusus untuk penyakit Sandhoff. Seperti penyakit Tay-Sachs, perawatan suportif termasuk menjaga saluran udara tetap terbuka setiap saat, serta nutrisi dan hidrasi yang tepat. Antikonvulsan awalnya dapat mengontrol epilepsi (kejang).
penyakit Krabbe(juga dikenal sebagai leukodistrofi sel globular dan lipidosis galaktosilceramida) adalah penyakit resesif autosomal yang disebabkan oleh defisiensi enzim galaktoserebrosidase. Penyakit ini paling sering menyerang bayi dengan onset sebelum usia 6 bulan, tetapi dapat terjadi selama masa remaja atau dewasa.
Akumulasi lemak yang tidak tercerna mempengaruhi pertumbuhan selubung pelindung saraf (selubung mielin) dan menyebabkan kerusakan parah pada keterampilan mental dan motorik. Gejala lain termasuk kelemahan otot, penurunan kemampuan otot untuk meregang (hipotonia), ketegangan otot (spastisitas), kejutan mendadak atau kedutan pada anggota badan (kejang mioklonik), lekas marah, demam yang tidak dapat dijelaskan, tuli, kebutaan, kelumpuhan, dan kesulitan menelan. Penurunan berat badan jangka panjang juga bisa terjadi.
Penyakit ini dapat didiagnosis dengan pengujian enzim dan mengidentifikasi pengelompokan sel yang khas menjadi badan globoid di materi putih otak, demielinasi saraf dan degenerasi, dan penghancuran sel-sel otak. Pada bayi, penyakit ini biasanya berakibat fatal sebelum usia 2 tahun. Orang dengan bentuk penyakit yang lebih lanjut memiliki perjalanan penyakit yang lebih ringan dan hidup secara signifikan lebih lama.
Tidak ada pengobatan khusus untuk penyakit Krabbe yang telah dikembangkan, meskipun transplantasi sumsum tulang dini dapat membantu beberapa orang. Orang dengan bentuk penyakit yang lebih lanjut memiliki perjalanan penyakit yang lebih ringan dan hidup secara signifikan lebih lama.
Leukodistrofi metakromatik, atau MLD, adalah sekelompok gangguan yang ditandai dengan akumulasi zat lemak dalam materi putih sistem saraf pusat dan saraf perifer dan sampai batas tertentu di ginjal. Mirip dengan penyakit Krabbe, MLD mempengaruhi mielin, yang menutupi dan melindungi saraf. Gangguan resesif autosomal ini disebabkan oleh defisiensi enzim arilsulfatase A. Gangguan ini mempengaruhi baik pria maupun wanita.
Leukodistrofi metakromatik memiliki tiga bentuk karakteristik: bayi akhir, remaja, dan dewasa.
- MLD Bayi Terlambat biasanya dimulai antara 12 dan 20 bulan setelah lahir. Anak-anak mungkin tampak normal pada awalnya, tetapi mengalami kesulitan berjalan dan kecenderungan untuk jatuh, disertai dengan nyeri intermiten di lengan dan kaki. kehilangan penglihatan progresif yang menyebabkan kebutaan, keterlambatan perkembangan dan hilangnya pencapaian yang diperoleh sebelumnya, gangguan menelan, kejang, dan demensia hingga 2 bertahun-tahun. Anak-anak juga mengalami pengecilan otot dan kelemahan progresif dan akhirnya kehilangan kemampuan mereka untuk berjalan. Sebagian besar anak dengan bentuk kelainan ini meninggal pada usia 5 tahun.
- MLD Remaja biasanya dimulai antara usia 3 dan 10 tahun. Gejala termasuk gangguan kinerja sekolah, gangguan mental, ataksia, kejang, dan demensia. Gejala berkembang, dengan kematian terjadi 10-20 tahun setelah timbulnya penyakit.
- Bentuk dewasa. Gejala dewasa onset setelah usia 16 dan mungkin termasuk ataksia, kejang, tremor abnormal pada tungkai (tremor), gangguan konsentrasi, depresi, gangguan jiwa dan demensia. Kematian biasanya terjadi 6-14 tahun setelah timbulnya gejala.
MLD tidak dapat disembuhkan. Pengobatan bersifat simtomatik dan suportif. Transplantasi sumsum tulang dalam beberapa kasus dapat menunda perkembangan penyakit. Kemajuan yang signifikan telah dibuat dengan terapi gen pada model hewan dengan MLD dan dalam uji klinis.
penyakit Wolmanjuga dikenal sebagai defisiensi asam lisosom lipaseadalah gangguan penyimpanan lipid serius yang biasanya berakibat fatal pada usia 1 tahun. Gangguan resesif autosomal ini ditandai dengan akumulasi ester kolesterol (biasanya bentuk transportasi kolesterol) dan trigliserida (bentuk kimia di mana lemak ada di dalam tubuh), yang dapat menumpuk secara signifikan dan menyebabkan kerusakan sel dan kain.
Baik pria maupun wanita menderita gangguan ini. Bayi normal dan aktif saat lahir, tetapi dengan cepat mengalami gangguan mental progresif, hati yang membesar dan limpa yang sangat besar, kembung, masalah pencernaan. penyakit kuning, anemia, muntah dan deposit kalsium di kelenjar adrenalmenyebabkan mereka mengeras.
Tipe lain Defisiensi lipase asam lisosom adalah penyakit penyimpanan ester kolesterol. Penyakit yang sangat langka ini terjadi sebagai akibat dari penyimpanan ester kolesterol dan trigliserida dalam sel darah, limfe dan jaringan limfoid. Pada anak-anak di masa dewasa, hati membesar, yang menyebabkan sirosis dan kronis gagal hati. Anak-anak mungkin juga memiliki deposit kalsium di kelenjar adrenal, dan penyakit kuning dapat berkembang pada tahap akhir penyakit.
Saat ini, masalah penggantian enzim sedang dipelajari secara aktif baik pada penyakit Wolman maupun dalam akumulasi ester kolesterol.
Perhatian! Informasi ini hanya untuk tujuan informasi dan tidak dimaksudkan untuk mendiagnosis dan meresepkan pengobatan. Selalu konsultasikan dengan dokter spesialis Anda!