Lipoidosi (malattia da accumulo di lipidi lisosomiali): che cos'è, esempi
Attenzione! Le informazioni sono solo a scopo informativo e non hanno lo scopo di diagnosticare e prescrivere trattamenti. Consulta sempre il tuo medico specialista!
Contenuto
- Cosa sono le malattie da accumulo lisosomiale?
- Cosa sono i lipidi?
- Come si eredita la lipoidosi?
- Tipi e sintomi delle malattie da accumulo lisosomiale
Cosa sono le malattie da accumulo lisosomiale?
Malattie da accumulo lisosomiale (lipoidosi), sono un gruppo di malattie metaboliche ereditarie in cui quantità dannose di sostanze grasse (lipidi) si accumulano in varie cellule e tessuti del corpo.
Le persone con questi disturbi o non producono abbastanza di uno degli enzimi necessari per abbattere (metabolizzare) i lipidi o producono enzimi che non funzionano correttamente.
Nel tempo, questo eccessivo accumulo di grasso può portare a danni permanenti a cellule e tessuti, soprattutto nel cervello, sistema nervoso periferico (nervi dal midollo spinale al resto del corpo), fegato, milza e midollo osseo.
Cosa sono i lipidi?
I lipidi sono sostanze simili ai grassi che sono parti importanti delle membrane all'interno e tra le cellule e nella guaina mielinica, che copre e protegge i nervi. I lipidi includono oli, acidi grassi, cere, steroidi (come colesterolo ed estrogeni) e altri composti correlati.
Queste sostanze grasse sono immagazzinate naturalmente nelle cellule, negli organi e nei tessuti del corpo. Piccoli corpi all'interno delle cellule, chiamati lisosomi, convertono o metabolizzano regolarmente lipidi e proteine in componenti più piccoli per fornire energia al corpo. I disturbi in cui il materiale intracellulare, che non può essere metabolizzato, rimane nei lisosomi, sono chiamati malattie da accumulo lisosomiale.
Oltre alle malattie associate all'accumulo di lipidi, altre malattie lisosomiali associate all'accumulo includono le mucolipidosi, in cui si accumulano cellule e tessuti una quantità eccessiva di lipidi con molecole di zucchero ad essi attaccate, nonché mucopolisaccaridosi, in cui si accumula una quantità eccessiva di zuccheri grandi e complessi molecole.
Come vengono ereditati? lipoidosi?
Le malattie da accumulo lisosomiale sono ereditate da uno o entrambi i genitori portatori di un gene difettoso che regola uno specifico enzima che metabolizza i lipidi in una classe di cellule del corpo. Possono essere ereditati in due modi:
- Eredità autosomica recessiva si verifica quando entrambi i genitori portano e trasmettono una copia del gene difettoso, ma nessuno dei due è affetto dal disturbo. Ogni bambino nato da questi genitori ha il 25% di possibilità di ereditare entrambe le copie del gene difettoso, il 50% la probabilità che sia un portatore simile ai suoi genitori e una probabilità del 25% di non ereditare alcuna copia del difettoso gene. I bambini di entrambi i sessi possono essere ereditati con modalità autosomica recessiva.
- Eredità recessiva legata all'X (o correlata al sesso) si verifica quando la madre porta il gene affetto sul cromosoma X. I cromosomi X e Y sono coinvolti nella determinazione del sesso. Le donne hanno due cromosomi X, mentre gli uomini hanno un cromosoma X e un cromosoma Y. I figli di femmine portatrici hanno una probabilità del 50% di ereditare e influenzare la malattia, poiché i figli ricevono un cromosoma X dalla madre e un cromosoma Y dal padre. Le figlie hanno una probabilità del 50% di ereditare il cromosoma X interessato dalla madre e sono portatrici o lievemente affette. Gli uomini affetti non trasmettono la malattia ai loro figli, ma le loro figlie saranno portatrici e portatrici della malattia.
Tipi e sintomi delle malattie da accumulo lisosomiale
malattia di Gaucher - la più comune delle malattie lisosomiali. La malattia è causata da una carenza dell'enzima glucocerebrosidasi, che porta all'accumulo di glucocerebroside in molti tessuti. Il materiale grasso può accumularsi nel cervello, nella milza, nel fegato, nei reni, nei polmoni e nel midollo osseo.
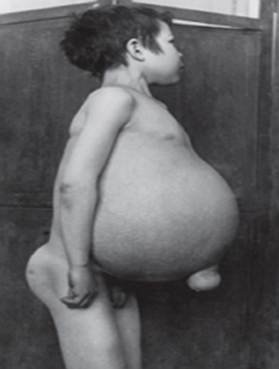
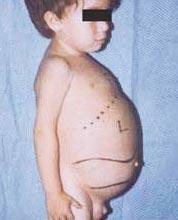
I sintomi possono includere danni al cervello, ingrossamento della milza e fegato, funzionalità epatica compromessa, disturbi scheletrici e danni ossei che possono causare dolore e fratture, gonfiore dei linfonodi e (a volte) delle articolazioni adiacenti, gonfiore, carnagione brunastra, anemia, basso numero di piastrine e macchie gialle sugli occhi.
Gli individui più gravemente colpiti possono anche essere più suscettibili alle infezioni. La malattia colpisce allo stesso modo uomini e donne.
La malattia di Gaucher ha tre sottotipi clinici generali:
- Tipo 1 (O no neuronopatico tipo) è la forma più comune della malattia negli Stati Uniti e in Europa. Il cervello non è interessato, ma possono verificarsi danni ai polmoni e, in rari casi, ai reni. I sintomi possono iniziare presto nella vita o nell'età adulta e comprendono un ingrossamento del fegato e un grave ingrossamento della milza, che possono portare a rotture e ulteriori complicazioni. La debolezza scheletrica e la malattia ossea possono essere estese. Le persone in questo gruppo di solito si contraggono facilmente a causa del basso numero di piastrine nel sangue. Possono anche provare affaticamento a causa dell'anemia. A seconda dell'insorgenza e della gravità della malattia, le persone con tipo 1 può vivere fino all'età adulta. Molti malati hanno una malattia lieve o potrebbero non mostrare alcun sintomo. Benchè 1genere La malattia di Gaucher è comune tra le persone di origine ebraica ashkenazita e può colpire persone di tutte le etnie.
- Tipo 2 (o neuropatico infantile acuto malattia di Gaucher) di solito inizia entro 3 mesi dalla nascita. I sintomi includono danni cerebrali estesi e progressivi, spasticità, convulsioni, rigidità degli arti, ingrossamento del fegato (epatomegalia epatica) e milza (splenomegalia), movimento oculare anomalo e scarsa capacità di succhiare e deglutire. I bambini affetti di solito muoiono prima dei 2 anni di età.
- Tipo 3 (neuronopatico cronico modulo) può iniziare in qualsiasi momento durante l'infanzia o anche in età adulta. È caratterizzata da sintomi neurologici lentamente progressivi ma meno gravi rispetto alla malattia di Gaucher acuta 2 tipi. I sintomi principali includono disturbi del movimento oculare, deficit cognitivi, scarsa coordinazione, convulsioni, ingrossamento della milza e/o del fegato, disturbi scheletrici, disturbi del sangue inclusa anemia e problemi con respirazione. Quasi tutti con la malattia di Gaucher 3 tipi, chi riceve la terapia enzimatica sostitutiva raggiungerà l'età adulta.
Per i pazienti 1° e 3° tipo La terapia enzimatica sostitutiva, somministrata per via endovenosa ogni due settimane, può ridurre drasticamente le dimensioni del fegato e della milza, ridurre le anomalie scheletriche e far regredire altre manifestazioni. Un trapianto di midollo osseo di successo tratta le manifestazioni neurologiche della malattia. Tuttavia, questa procedura comporta rischi significativi e viene eseguita raramente in soggetti con malattia di Gaucher.
In rari casi, può essere necessario un intervento chirurgico per rimuovere tutta o parte della milza (se una persona ha una conta piastrinica molto bassa o quando un organo ingrossato influisce notevolmente sul comfort della persona). La trasfusione di sangue può giovare ad alcune persone con anemia. Altri potrebbero aver bisogno di un intervento chirurgico di sostituzione dell'articolazione per migliorare la mobilità e la qualità della vita. Attualmente non esiste un trattamento efficace per il danno cerebrale che può verificarsi nelle persone con malattia di Gaucher di tipo 2 e 3.
Malattia di Niemann-Pick è un gruppo di malattie autosomiche recessive causate dall'accumulo di grasso e colesterolo nelle cellule del fegato, della milza, del midollo osseo, dei polmoni e, in alcuni casi, del cervello.
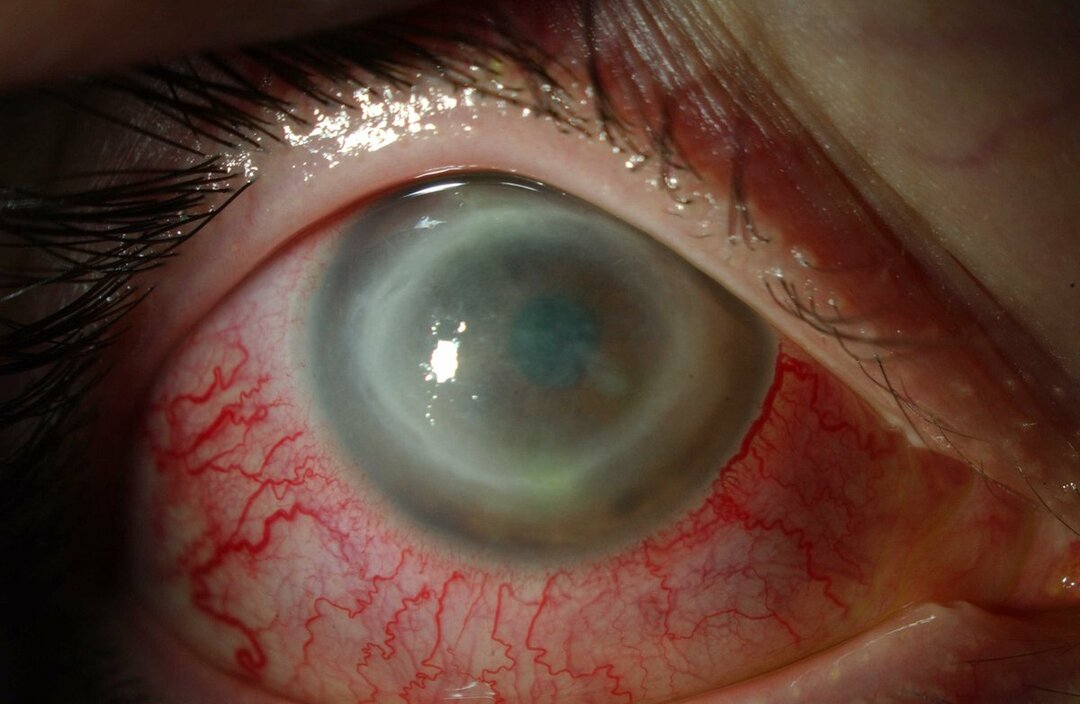
Le complicanze neurologiche possono includere atassia (mancanza di coordinazione muscolare che può influenzare il camminare, scrivere e mangiare, tra le altre funzioni), paralisi oculare, degenerazione cerebrale, problemi di apprendimento, spasticità, difficoltà di alimentazione e deglutizione, linguaggio, perdita del tono muscolare, ipersensibilità al tatto e una certa opacità della cornea dovuta all'eccessivo accumulo materiali. Nel 50% delle persone colpite si sviluppa un caratteristico alone rosso ciliegia intorno al centro della retina, visibile da un medico con uno strumento apposito.
La malattia di Niemann-Pick è classificata in tre sottotipi generali:
- Digitare un, la forma più grave, inizia nella prima infanzia. I bambini sembrano normali alla nascita, ma entro 6 mesi si sviluppa un danno cerebrale profondo, fegato e milza ingrossati e linfonodi e linfonodi sottocutanei ingrossati (xantomi). La milza può diventare 10 volte la sua dimensione normale e può rompersi, causando sanguinamento. Questi bambini diventano progressivamente più deboli, perdono la funzione motoria, possono diventare anemici e sono soggetti a infezioni ricorrenti. Raramente vivono più di 18 mesi. Questa forma della malattia è più comune nelle famiglie ebree.
- Tipo B(o esordio giovanile) di solito non colpisce il cervello, ma la maggior parte dei bambini si sviluppa atassia, danni ai nervi in uscita dal midollo spinale (neuropatia periferica), e difficoltà polmonari che progrediscono con l'età. L'ingrossamento del fegato e della milza è tipico degli adolescenti. Persone con tipo B possono vivere relativamente a lungo, ma molti richiedono ossigenoterapia per tutta la vita a causa di danni ai polmoni. I tipi Niemann-Pick A e B sono il risultato di un accumulo di una sostanza grassa chiamata sfingomielina a causa di una carenza di un enzima chiamato sfingomielinasi.
- Tipo C può apparire presto nella vita o svilupparsi durante l'adolescenza o addirittura l'età adulta. Malattia di Niemann-Picktipo C è causato non dal deficit di sflingomielinasi, ma da una mancanza di proteine NPC1 o NPC2. Di conseguenza, vari lipidi e in particolare il colesterolo si accumulano nelle cellule nervose e causano il loro malfunzionamento. Il coinvolgimento del cervello può essere esteso, con conseguente incapacità di guardare in alto e in basso, difficoltà a camminare e deglutire, perdita progressiva dell'udito e progressiva demenza. Persone con tipo C hanno solo un lieve ingrossamento della milza e del fegato. Aspettativa di vita delle persone con tipo C varia notevolmente. Alcune persone muoiono durante l'infanzia, mentre altre che sono meno colpite possono anche vivere in età adulta.
Attualmente non esiste una cura per la malattia di Niemann-Pick. Il trattamento è di supporto. I bambini di solito muoiono per infezione o perdita neurologica progressiva. Ci sono stati casi di trapianto di midollo osseo in diversi pazienti con tipo B con risultati contrastanti.
malattia di Fabryconosciuto anche come deficit di alfa-galattosidasi-A, provoca l'accumulo di materia grassa nel sistema nervoso autonomo (quella parte del sistema nervoso che controlla le funzioni involontarie come la respirazione e il battito cardiaco), negli occhi, nei reni e nel sistema cardiovascolare sistema.
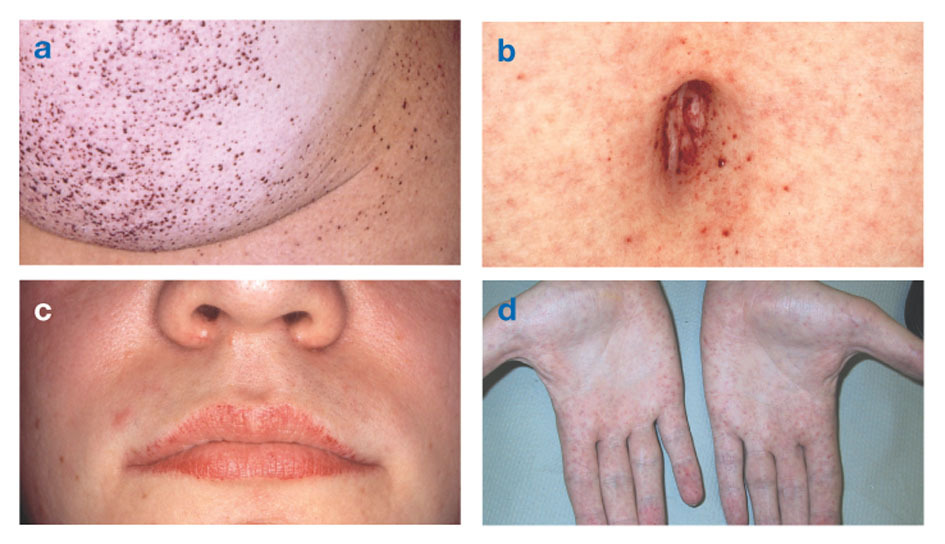
La malattia di Fabry è l'unica malattia lipidica legata all'X. La malattia colpisce principalmente gli uomini, anche se nelle donne si manifesta una forma più lieve e variabile. Raramente, le donne affette hanno sintomi gravi simili a quelli osservati negli uomini con il disturbo. L'insorgenza dei sintomi di solito si verifica durante l'infanzia o l'adolescenza.
I segni neurologici includono dolore bruciante alle braccia e alle gambe che peggiora con il caldo o dopo esercizio fisico, nonché l'accumulo di materiale in eccesso negli strati trasparenti della cornea (che porta all'opacizzazione, ma senza alterazioni della vista). L'accumulo di grasso nelle pareti dei vasi sanguigni può interrompere la circolazione, mettendo a rischio una persona ictus o attacco di cuore. Altri sintomi includono un cuore ingrossato, progressivo insufficienza renaleportando a malattia renale allo stadio terminale, problemi gastrointestinali, diminuzione della sudorazione e febbre.
Gli angiocheratomi (piccole chiazze in rilievo sulla pelle, non cancerose, rosso-violacee) possono svilupparsi nella parte inferiore del tronco e diventare più comuni con l'età.
Le persone con la malattia di Fabry spesso muoiono prematuramente per complicazioni dovute a insufficienza cardiaca, insufficienza renale o ictus. Farmaci come la fenitoina e la carbamazepina vengono spesso prescritti per trattare il dolore che accompagna la malattia di Fabry, ma non per curarlo. La metoclopramide o il Lipisorb (integratore alimentare) possono alleviare i disturbi gastrointestinali che si verifica spesso nelle persone con malattia di Fabry e alcune persone potrebbero aver bisogno di un trapianto di rene o dialisi. La sostituzione enzimatica può ridurre lo stoccaggio, alleviare il dolore e preservare la funzione degli organi in alcune persone con malattia di Fabry.
La malattia di Farber conosciuto anche come lipogranulomatosi di Farber, descrive un gruppo di rare malattie autosomiche recessive che causano l'accumulo di materia grassa nelle articolazioni, nei tessuti e nel sistema nervoso centrale. Colpisce sia gli uomini che le donne. La malattia di solito inizia nella prima infanzia, ma può manifestarsi più tardi nella vita.
I bambini con la classica malattia di Farber si sviluppano sintomi neurologici, che possono includere aumento della sonnolenza e letargia e problemi di deglutizione. Anche il fegato, il cuore e i reni possono essere colpiti. Altri sintomi possono includere contratture articolari (accorciamento cronico di muscoli o tendini intorno alle articolazioni), vomito, artrite, ingrossamento linfonodi, infiammazione articolare, raucedine e noduli sottocutanei che si addensano intorno alle articolazioni man mano che progredisce malattie.
Le persone affette con difficoltà respiratorie potrebbero aver bisogno di inserire un tubo di respirazione. La maggior parte dei bambini con questa condizione muore all'età di 2 anni, di solito da malattie polmonari. In una delle forme più gravi della malattia, poco dopo la nascita possono essere diagnosticati un ingrossamento del fegato e della milza. I bambini nati con questa forma della malattia di solito muoiono entro 6 mesi.
La malattia di Farber è causata da una carenza di un enzima chiamato ceramidasi. Attualmente non esiste un trattamento specifico per la malattia di Farber. I corticosteroidi possono essere prescritti per alleviare il dolore. Il trapianto di midollo osseo può ridurre i granulomi (piccole masse di tessuto infiammato) in persone con infiammazione lieve o in pazienti senza complicazioni polmonari o del sistema nervoso. Nelle persone anziane, i granulomi possono essere ridotti o rimossi chirurgicamente.
gangliosidosi comprende due diversi gruppi di malattie genetiche. Entrambi sono autosomici recessivi e colpiscono allo stesso modo maschi e femmine.
Gangliosidosi GM1.
La gangliosidosi GM1 è causata da un deficit dell'enzima beta-galattosidasi, con conseguente accumulo anomalo di materiali lipidici acidi, in particolare nelle cellule nervose del sistema nervoso centrale e periferico sistemi. La gangliosidosi GM1 ha tre manifestazioni cliniche:
- GM1 (il sottotipo più grave, che si verifica subito dopo la nascita) può includere neurodegenerazione, convulsioni, ingrossamento del fegato e della milza, ruvidità delle caratteristiche facciali, irregolarità scheletriche, rigidità articolare, gonfiore, debolezza muscolare, risposta esagerata allo spavento e problemi con andatura. Circa la metà delle persone colpite sviluppa macchie rosso ciliegia negli occhi. I bambini possono essere sordi e ciechi all'età di 1 anno e spesso muoiono all'età di 3 anni per complicazioni cardiache o polmonite.
- bambino in ritardo La gangliosidosi GM1 di solito inizia tra 1 e 3 anni. I sintomi neurologici includono atassia, convulsioni, demenza e difficoltà a parlare.
- Gangliosidosi GM1 si sviluppa tra i 3 e i 30 anni. I sintomi includono diminuzione della massa muscolare (atrofia muscolare), complicanze neurologiche che sono meno gravi e progrediscono più lentamente rispetto ad altre forme disturbi, opacità corneale in alcune persone e contrazioni muscolari prolungate che causano movimenti di torsione e ripetitivi o posture anomale (distonia). Gli angiocheratomi possono svilupparsi nella parte inferiore del tronco del corpo. La dimensione del fegato e della milza è normale nella maggior parte delle persone colpite.
Gangliosidosi GM2.
La gangliosidosi GM2 induce anche il corpo ad accumulare sostanze grasse acide in eccesso nei tessuti e nelle cellule, principalmente nelle cellule nervose. Questi disturbi derivano da una carenza dell'enzima beta-esosaminidasi. I disturbi GM2 includono:
- malattia di Tay-Sachs (nota anche come gangliosidosi GM2 variante B) e le sue forme varianti sono causate da un deficit dell'enzima esosaminidasi A. I bambini affetti si sviluppano normalmente durante i primi mesi di vita. I sintomi iniziano a 6 mesi di età e comprendono perdita progressiva della capacità mentale, demenza, diminuzione del contatto visivo, aumento una risposta sbalorditiva al rumore, perdita progressiva dell'udito che porta a sordità, difficoltà a deglutire, cecità, macchie rosso ciliegia sulla retina e alcuni paralisi. Le convulsioni possono iniziare nel secondo anno di vita di un bambino. I bambini possono eventualmente diventare dipendenti dal tubo di alimentazione e spesso muoiono all'età di 4 anni a causa di infezioni ricorrenti. Sfortunatamente, non esiste un trattamento speciale. Gli anticonvulsivanti possono inizialmente controllare le convulsioni. Altri trattamenti di supporto includono una corretta alimentazione e idratazione e metodi per mantenere aperte le vie aeree. Una forma rara del disturbo chiamato malattia di Tay-Sachs ad esordio tardivo, si verifica nelle persone di età compresa tra 20 e 30 anni ed è caratterizzato da un'andatura instabile e un progressivo deterioramento neurologico.
- La malattia di Sandhoff (Opzione AB) è una forma grave della malattia di Tay-Sachs. L'esordio di solito si verifica a 6 mesi di età e non è limitato a nessun gruppo etnico. I segni neurologici possono includere un progressivo deterioramento del sistema nervoso centrale, motorio debolezza, cecità precoce, grave reazione di paura al suono, spasticità, shock o spasmi muscoli (mioclono), epilessia, una testa ingrandita in modo anomalo (macrocefalia) e macchie rosse negli occhi. Altri sintomi possono includere frequenti infezioni respiratorie, soffio cardiaco, tratti del viso da bambola e ingrossamento del fegato e della milza. Non esiste un trattamento specifico per la malattia di Sandhoff. Come per la malattia di Tay-Sachs, l'assistenza di supporto include il mantenimento delle vie aeree sempre aperte, nonché una corretta alimentazione e idratazione. Gli anticonvulsivanti possono inizialmente controllare l'epilessia (convulsioni).
Malattia di Krabbe(conosciuto anche come leucodistrofia a cellule globulari e lipidosi galattosilceramide) è una malattia autosomica recessiva causata da un deficit dell'enzima galattocerebrosidasi. La malattia colpisce più comunemente i neonati con esordio prima dei 6 mesi di età, ma può manifestarsi durante l'adolescenza o l'età adulta.
L'accumulo di grassi non digeriti influisce sulla crescita della guaina protettiva del nervo (guaina mielinica) e provoca una grave compromissione delle capacità mentali e motorie. Altri sintomi includono debolezza muscolare, ridotta capacità dei muscoli di allungarsi (ipotonia), tensione muscolare (spasticità), shock improvviso o contrazioni degli arti (crisi miocloniche), irritabilità, febbre inspiegabile, sordità, cecità, paralisi e difficoltà a deglutire. Può anche verificarsi una perdita di peso a lungo termine.
La malattia può essere diagnosticata mediante test enzimatici e identificando il suo caratteristico raggruppamento cellulare in corpi globoidi nella sostanza bianca del cervello, demielinizzazione dei nervi e degenerazione e distruzione delle cellule cerebrali. Nei neonati, la malattia è solitamente fatale prima dei 2 anni di età. Le persone con una forma più avanzata della malattia hanno un decorso più lieve della malattia e vivono significativamente più a lungo.
Non è stato sviluppato alcun trattamento specifico per la malattia di Krabbe, sebbene il trapianto di midollo osseo precoce possa aiutare alcune persone. Le persone con una forma più avanzata della malattia hanno un decorso più lieve della malattia e vivono significativamente più a lungo.
Leucodistrofia metacromatica, o DLM, è un gruppo di disturbi caratterizzati dall'accumulo di sostanze grasse nella sostanza bianca del sistema nervoso centrale e nei nervi periferici e in una certa misura nei reni. Simile alla malattia di Krabbe, la MLD colpisce la mielina, che copre e protegge i nervi. Questa malattia autosomica recessiva è causata da un deficit dell'enzima arilsulfatasi A. Questo disturbo colpisce sia gli uomini che le donne.
La leucodistrofia metacromatica ha tre forme caratteristiche: lattante tardivo, giovanile e adulto.
- Late Infant MLD di solito inizia tra 12 e 20 mesi dopo la nascita. I bambini possono inizialmente sembrare normali, ma hanno difficoltà a camminare e tendenza a cadere, accompagnati da dolore intermittente alle braccia e alle gambe. perdita progressiva della vista che porta a cecità, ritardi nello sviluppo e perdita di traguardi precedentemente acquisiti, deglutizione ridotta, convulsioni e demenza fino a 2 anni. I bambini sviluppano anche atrofia muscolare progressiva e debolezza e alla fine perdono la capacità di camminare. La maggior parte dei bambini con questa forma del disturbo muore all'età di 5 anni.
- MLD giovanile di solito inizia tra i 3 ei 10 anni. I sintomi includono prestazioni scolastiche ridotte, menomazione mentale, atassia, convulsioni e demenza. I sintomi progrediscono, con la morte che si verifica 10-20 anni dopo l'inizio della malattia.
- Forma adulta. Sintomi adulti esordio dopo i 16 anni e può includere atassia, convulsioni, tremori anormali degli arti (tremori), concentrazione ridotta, depressione, disturbi mentali e demenza. La morte di solito si verifica 6-14 anni dopo l'insorgenza dei sintomi.
La MLD non può essere curata. Il trattamento è sintomatico e di supporto. Il trapianto di midollo osseo può in alcuni casi ritardare la progressione della malattia. Sono stati compiuti progressi significativi con la terapia genica in modelli animali con MLD e negli studi clinici.
Malattia di Wolmanconosciuto anche come deficit di lipasi acida lisosomialeè una grave malattia da accumulo di lipidi che di solito è fatale a 1 anno di età. Questa malattia autosomica recessiva è caratterizzata da un accumulo di esteri del colesterolo (di solito la forma di trasporto del colesterolo) e trigliceridi (la forma chimica in cui esistono i grassi nel corpo), che possono accumularsi in modo significativo e causare danni alle cellule e tessuti.
Sia gli uomini che le donne soffrono di questo disturbo. I neonati sono normali e attivi alla nascita, ma sviluppano rapidamente una compromissione mentale progressiva, un fegato ingrossato e una milza notevolmente ingrossata, gonfiore, problemi gastrointestinali. ittero, anemia, vomito e depositi di calcio in ghiandole surrenalifacendoli indurire.
Un altro tipo Il deficit di lipasi acida lisosomiale è malattia da accumulo di esteri del colesterolo. Questa malattia estremamente rara si verifica a causa dell'accumulo di esteri del colesterolo e trigliceridi nelle cellule del sangue, linfa e tessuto linfoide. Nei bambini in età adulta, il fegato si ingrandisce, il che porta alla cirrosi e cronico insufficienza epatica. I bambini possono anche avere depositi di calcio nelle ghiandole surrenali e l'ittero può svilupparsi nelle fasi successive della malattia.
Attualmente, il problema della sostituzione enzimatica viene attivamente studiato sia nella malattia di Wolman che nell'accumulo di un estere del colesterolo.
Attenzione! Le informazioni sono solo a scopo informativo e non hanno lo scopo di diagnosticare e prescrivere trattamenti. Consulta sempre il tuo medico specialista!