Robinova sindroms: kas tas ir, cēloņi, pazīmes, ārstēšana, prognoze
Saturs
- Kas ir Robinofa sindroms?
- Robinova sindroma klīniskās pazīmes
- Robinova sindroma cēloņi
- Ietekmētās populācijas
- Diagnostika
- Robinova sindroma ārstēšana
- Prognoze
- Līdzīgi traucējumi
Kas ir Robinofa sindroms?
Robinofa sindroms ir ārkārtīgi reta iedzimta slimība, kas ietekmē kaulu un citu ķermeņa daļu attīstību. Pastāv divas Robinova sindroma formas, kas atšķiras pēc pazīmēm un simptomiem, smaguma pakāpes, mantojuma veida un ar tiem saistītajiem gēniem.
Vispirms, autosomāli recesīvs Robinova sindroms ir smagāks, un to raksturo:
- garu kaulu saīsināšana rokās un kājās;
- īsi pirksti un kāju pirksti;
- mugurkaula mugurkaula formas kauli, kas noved pie patoloģiska mugurkaula izliekuma (kifoskolioze);
- iztukšo vai trūkst ribu;
- īss augums;
- atšķirīgas sejas īpašības.
Citas funkcijas var ietvert:
- dzimumorgānu nepietiekama attīstība;
- zobu problēmas;
- nieru vai sirds defekti;
- attīstības kavēšanās.
Bērni ar otrā forma, autosomāli dominējošajam Robinova sindromam ir līdzīgi simptomi, bet tas izpaužas maigāk. Mugurkaula un ribu anomālijas parasti nav, un īss augums ir mazāk izteikts.
Dažiem cilvēkiem ar autosomāli dominējošo Robinova sindromu var būt arī palielināts kaulu minerālais blīvums (osteoskleroze).
Robinova sindroma klīniskās pazīmes

Robinova autosomāli recesīvo sindromu raksturo:
- īss augums;
- raksturīgās sejas īpašības;
- skeleta patoloģijas un / vai dzimumorgānu patoloģijas.
Klīnisko pazīmju klāsts un smagums dažādiem cilvēkiem ir atšķirīgs.
Lielākajai daļai bērnu ar Robinofa sindromu ir pieredze augšanas aizkavēšanās pēc piedzimšanaskā rezultātā augums ir mazs vai vidēji īss. Lielākajai daļai bērnu ir normāls intelekts, bet aptuveni 20 procentiem skarto personu var būt:
- garīgi traucējumi;
- kavēšanās sasniegt starpposma mērķus;
- kavēšanās valodu prasmju attīstībā.
Sejas vaibsti atgādina augļa attīstības pazīmesMedicīnas literatūrā to bieži dēvē par "augļa seju". Raksturīgas galvas un sejas zonas anomālijas var ietvert liela galva (makrocefālija) ar izliektu pieri un vidējās virsmas nepietiekama attīstība (vidējās virsmas hipoplāzija).
Ietekmētiem bērniem var būt arī:
- Plaši izvietotas acis (acu hipertelorisms), kas izvirzās no orbītām (izliektas acis)
- neparasti plaši slīpi plakstiņi (acu spraugas);
- mazs augšupvērsts deguns ar nāsīm, kas uzliesmoja uz priekšu;
- nogrimis priekšgala tilts;
- Nepareizi novietotas (t. I., Zemas, pagrieztas atpakaļ) ausis.
Dažiem skartajiem bērniem var būt:
- plata, trīsstūrveida mute, pagriezta uz leju, ar augšējās lūpas centrā garu šķēlumu;
- mazs zods;
- mazs žoklis (mikrognātija);
- smaganu hiperplāzija.
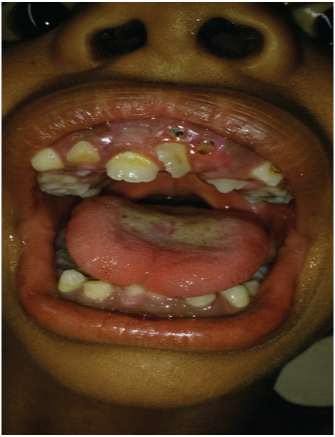
Var būt arī zobu patoloģijas, tostarp:
- nepareiza zobu novietošana;
- zobu saspiešana aizmugurē;
- kavēta sekundāro (pastāvīgo) zobu izvirdums.
Mīksto audu struktūra rīkles aizmugurē (uvula) var būt nepietiekami attīstīta vai nenormāli sadalīta (divdaļīga). Skartiem bērniem var būt arī aukslēju šķeltne, neparasta vertikāla grope vai caurums augšlūpā (lūpas šķeltne) un / vai ierobežota mēles kustība (ankiloglosija).
Ankiloglosija var veicināt kavēšanos valodu prasmju attīstībā. Lielākajai daļai bērnu ar Robinova sindromu saistītās sejas anomālijas ar vecumu kļūst mazāk izteiktas.
Skeleta patoloģijas var ietvert:
- apakšdelma kaulu patoloģijas (rādiuss un elkoņa kauls), kas izskatās neparasti īsi un nepietiekami attīstīti (apakšdelma brahimēlija);
- neparasta apakšdelma īkšķa sānu novirze (rādiuss) rādiusa saīsināšanās dēļ (plaukstas locītava);
- neparasti īsi pirksti (brachydactyly);
- pastāvīga piektā pirksta fiksācija saliektā stāvoklī (klinodactyly);
- neparasti mazas rokas ar platiem īkšķiem.
Lielo pirkstu gala kauli (gala falangas) var būt nenormāli atdalīti un / vai pirkstu un pirkstu kauli (falangas) var būt nepietiekami attīstīti (hipoplastiski). Papildu novirzes var ietvert:
- gūžas locītavas pārvietošana;
- ierobežots elkoņa pagarinājums, patoloģiska saplūšana vai noteiktu ribu trūkums;
- patoloģisks mugurkaula izliekums no vienas puses uz otru (skolioze);
- kaulu (skriemeļu) vienas puses nepietiekama attīstība mugurkaula (hemivertebra) vidējā (krūšu) daļā;
- dažu skriemeļu saplūšana.
Lasiet arī:Ehlers-Danlos sindroms
Ietekmētie bērni var piedzīvot patoloģiska kaulu padziļināšanāsveidojot krūškurvja centru ("piltuves lāde"). Bērniem var būt arī deformēti (displastiski) nagi un / vai anomālijas pirkstu un plaukstu ādas kores rakstos.
Lielākajai daļai bērnu ar Robinofa sindromu ir arī dzimumorgānu anomālijasun dažiem var būt ārēji dzimumorgāni, kas nav skaidri vīrieši vai sievietes (neskaidri dzimumorgāni). Dzimumu parasti var pareizi noteikt agrā bērnībā. Sieviešu vidū Klitoris un ārējās iegarenās ādas krokas abās maksts atveres pusēs (labia majora) var būt nepietiekami attīstītas (hipoplastiskas). Vīriešiem dzimumloceklis var būt neparasti mazs (mikropenis) un var būt paslēpts zem apkārtējās ādas; Turklāt viens vai abi sēklinieki nedrīkst nolaisties sēkliniekos (kriptorichisms).
Retos gadījumos skartajiem vīriešiem var būt patoloģiski zems sēklinieku funkcijas līmenis (daļēji primārais) hipogonādisms, bet ir normāla sekundāro seksuālo īpašību attīstība (piemēram, raupja balss, raksturīgi modeļi matu augšana, pēkšņs sēklinieku un sēklinieku augšanas un attīstības pieaugums utt.), izņemot noturību mikropenis.
Skartās sievietes uzrāda normālu olnīcu darbību (dzimumdziedzeru funkciju) un normālu auglību.
Cilvēkiem ar Robinofa sindromu var būt papildu fiziski traucējumi, piemēram:
- nieru dubultošanās;
- neparasta urīna uzkrāšanās nierēs (hidronefroze);
- resnās zarnas daļu izliekumi caur patoloģisku vēdera gļotādas atveri (cirkšņa trūce);
- resnās zarnas sekciju izspiedumi caur vēdera sienu pie nabas (nabas trūce).
Turklāt aptuveni 13 procentiem mazuļu ar Robinofa sindromu piedzimst sirds defekti (iedzimti sirds defekti). Šajos gadījumos visbiežākais sirds defekts ir "labā kambara obstrukcija", kurā tiek bloķēta asins plūsma no sirds apakšējās kameras (ventrikula). trauka patoloģiska sašaurināšanās (stenozes) vai slēgšanas (atrezijas) dēļ, kas rodas no kambara (plaušu stumbra) un sadalās kreisajā un labajā plaušā artērijas.
Simptomi, kas saistīti ar šo sirds defektu, ievērojami atšķiras atkarībā no šķēršļa lieluma un atrašanās vietas. Dažiem bērniem ar Robinofa sindromu var būt citas sirds problēmas, tostarp sarežģīti iedzimti sirds defekti, kas var izraisīt dzīvībai bīstamas komplikācijas.
Retos gadījumos zīdaiņi un bērni ar Robinofa sindromu var būt pakļauti atkārtotām plaušu infekcijām (pneimonija). Retos, smagos gadījumos bez atbilstošas ārstēšanas pneimonija var izraisīt dzīvībai bīstamas komplikācijas.
Dominējošajām un recesīvajām Robinova sindroma formām ir daudz tādu pašu simptomu un fizisko pazīmes (piemēram, galvaskausa -sejas patoloģijas, īss augums, skeleta malformācijas un dzimumorgānu hipoplāzija orgāni). Tomēr simptomi un fiziskās pazīmes, kas saistītas ar recesīvo formu, mēdz būt smagākas.
Zīdaiņiem ar recesīvo Robinova sindromu ir vairāk ribu anomāliju (piemēram, patoloģiska pārvietošana, saplūšana) un / vai noteiktu ribu trūkums) un defekti, kas ietekmē mugurkaula kaulus (skriemeļus) nekā dominējošiem zīdaiņiem slimības. Turklāt īsāks augums, apakšdelma kaulu nepietiekama attīstība (radiācijas hipoplāzija) un pirkstu anomālijas ir smagākas. Skartiem bērniem var būt viena no apakšdelma kauliem galvas mežģījums, un šī anomālija reti sastopama cilvēkiem ar Robinova sindroma dominējošo formu. Cilvēkiem ar recesīvu formu mutes forma var būt arī trīsstūrveida.
Autosomāli dominējošā Robinova sindroma variantam, osteosklerozes formai, ir raksturīga:
- palielināts kaulu minerālu blīvums, īpaši galvaskausā;
- normāla augšana;
- liela galva;
- un dzirdes zudums, papildus raksturīgajām pazīmēm un simptomiem.
Lasiet arī:Acetons bērna urīnā (acetonūrija bērniem): ko darīt, cēloņi, simptomi, ārstēšana
Robinova sindroma cēloņi

Autosomāli recesīvās un autosomāli dominējošās Robinova sindroma formas izraisa dažādu gēnu izmaiņas (mutācijas).
Autosomāli recesīvs veids slimība rodas gēna mutāciju dēļ ROR2kas noved pie olbaltumvielu trūkuma ROR2. Bez šī proteīna tiek traucēta bērna attīstība, īpaši skeleta, sirds un dzimumorgānu veidošanās.
Autosomāli dominējošais tips slimība rodas gēna mutāciju dēļ WNT5A vai gēns DVL1, kā rezultātā trūkst specifisku proteīnu, kas nepieciešami normālai attīstībai. Osteosklerozes formu izraisa mutācijas gēns DVL1.
Nelielai daļai bērnu ar Robinofa sindromu nevienā no šiem gēniem nav mutāciju, un slimības cēlonis joprojām nav zināms.
Lielāko daļu ģenētisko slimību nosaka divu gēnu kopiju statuss, vienu no tēva un otru no mātes. Recesīvi ģenētiski traucējumi rodas, ja persona manto divas neparasta gēna kopijas par vienu un to pašu pazīmi, vienu no katra vecāka. Ja persona manto vienu normālu gēnu un vienu slimības gēnu, persona pārnēsā šo slimību, bet parasti nerada simptomus. Risks diviem nesējiem vecākiem, kuri abi nodod mainīto gēnu un inficē bērnu, ir 25% ar katru grūtniecību. Risks iegūt bērnu, kurš būs nēsātājs, tāpat kā vecāks, ar katru grūtniecību ir 50%. Bērnam ir 25% iespēja iegūt normālus gēnus no abiem vecākiem. Risks vīriešiem un sievietēm ir vienāds.
Vecākiem, kas ir tuvi radinieki (incests), ir lielāka iespēja iegūt slimu bērnu nekā nesaistītiem vecākiem.
Dominējoši ģenētiski traucējumi rodas, ja ir nepieciešama tikai viena patoloģiska gēna kopija, lai izraisītu noteiktu slimību. Patoloģisko gēnu var mantot no jebkura vecāka, vai arī tas var būt jaunas mutācijas (gēnu maiņas) rezultāts skartajā personā. Risks pārnest patoloģisko gēnu no skartajiem vecākiem uz pēcnācējiem ir 50% katrai grūtniecībai. Risks vīriešiem un sievietēm ir vienāds.
Dažiem cilvēkiem šo traucējumu izraisa spontāna ģenētiska mutācija, kas rodas olšūnā vai spermā. Šādās situācijās traucējumi netiek mantoti no vecākiem.
Ietekmētās populācijas
Autosomāli recesīvs sindroms ir ziņots mazāk nekā 200 cilvēkiem ģimenēs no dažādām etniskajām grupām.
Ir ziņots par autosomāli dominējošo sindromu mazāk nekā 50 ģimenēs.
Diagnostika
Robinofa sindroma diagnoze parasti tiek veikta neilgi pēc piedzimšanas, pamatojoties uz fiziskiem konstatējumiem, tostarp īsu augumu, ekstremitāšu un dzimumorgānu anomālijām un sejas īpašībām.
Ir pieejama gēna molekulārā ģenētiskā pārbaude, lai apstiprinātu autosomāli recesīvā Robinova sindroma diagnozi ROR2. Gēnu mutāciju molekulārā ģenētiskā pārbaude WNT5A un gēns DVL1 pieejams, lai apstiprinātu autosomāli dominējoša slimības veida diagnozi.
Robinova sindroma ārstēšana
Robinova sindroma ārstēšana ir vērsta uz specifiskiem simptomiem, kas rodas katrai personai. Ārstēšanai var būt nepieciešama koordinēta speciālistu komanda. Pediatri, kaulu slimību ārstēšanas speciālisti (ortopēdi), ķirurgi, sirds problēmu diagnostikas un ārstēšanas speciālisti (kardiologiem), fizioterapeitiem un / vai citiem veselības aprūpes sniedzējiem var būt nepieciešama sistemātiska un visaptveroša ārstēšanas plānošana ievainots bērns.
Skartiem bērniem ar neskaidriem dzimumorgāniem dzimumu var pareizi noteikt, veicot klīnisko pārbaudi jaundzimušā periodā. Dažiem bērniem var veikt operāciju un / vai veikt citus pasākumus, lai izlabotu kriptorhidismu un / vai citas dzimumorgānu patoloģijas. Turklāt, lai uzlabotu dzimumlocekļa garumu un sēklinieku tilpumu, dažreiz tiek ievadīti hormonālie uztura bagātinātāji, piemēram, cilvēka horiona gonadotropīns un / vai testosterons. Ir konstatēts, ka dažiem pacientiem augšanas hormona deficīts pozitīvi reaģē uz augšanas hormona terapiju. Tomēr šīs hormonu terapijas formas rūpīgi jāuzrauga bērnu endokrinologam.
Īpaši vingrinājumi un / vai operācija var palīdzēt dažu mugurkaula traucējumu ārstēšanā. Operāciju var veikt arī, lai labotu cirkšņa trūces, noteiktas galvaskausa un sejas patoloģijas, smagu mugurkaula izliekumu (skoliozi), kas ir sekundāra saistībā ar anomālijām hemivertebrā un ribās, pilnībā vai daļēji savienotiem pirkstiem / kāju pirkstiem (sindaktiliski), lūpu / aukslēju šķeltni un / vai citiem defektiem attīstību. Bikšturi, zobu ķirurģija un / vai citas atbalstošas terapijas var palīdzēt labot nepareizi novietotus zobus un / vai citas mutes dobuma anomālijas.
Lasiet arī:Disleksija
Zīdaiņiem un bērniem ar Robinofa sindromu jāveic rūpīga medicīniskā pārbaude, lai nodrošinātu ātru sirds patoloģiju identificēšana un tūlītēja atbilstoša ārstēšana, kas varētu būt saistītas ar to traucējumi. Arī skartie zīdaiņi un bērni ir rūpīgi jāuzrauga, lai palīdzētu nekavējoties novērst un / vai atklāt plaušu infekcijas (pneimoniju) un nodrošināt tūlītēju, atbilstošu ārstēšanu.
Agrīna iejaukšanās ir būtiska, lai bērni ar Robinova sindromu sasniegtu savu potenciālu. Īpaši pakalpojumi, kas var būt noderīgi skartajiem bērniem, var ietvert ārstniecības līdzekļus izglītība, sociālais atbalsts, fizikālā terapija un / vai cita medicīniskā, sociālā un / vai profesionāli pakalpojumi.
Prognoze
Robinova sindroma prognoze parasti ir laba, taču sirdsdarbības traucējumu smagums var ietekmēt paredzamo dzīves ilgumu.
Līdzīgi traucējumi
Šādu traucējumu simptomi var būt līdzīgi Robinova sindromam. Diferenciāldiagnozei ir noderīgi salīdzinājumi:
AchondroplasiaIr iedzimts traucējums, kam raksturīgas neparasti īsas rokas un kājas un īss augums (īsu ekstremitāšu pundurisms), patoloģiskas sejas īpašības un / vai skeleta malformācijas. Sejas vaibsti var ietvert neparasti lielu galvu (makrocefālija), neparastu pieres izliekumu, zemu deguna tiltu un / vai vidējās sejas nepietiekamu attīstību (vidējās sejas hipoplāzija).
Skeleta anomālijas var ietvert neparasti īsus pirkstus un kāju pirkstus (brachydactyly), sagitālu mugurkaula līkumu, kas izvirzīts uz priekšu (lordoze), mugurkaula sašaurināšanās (stenoze). Papildu traucējumi var būt problēmas ar elkoņa un gūžas pagarinājumu, samazināts muskuļu tonuss (hipotensija) un / vai biežas vidusauss infekcijas (vidusauss iekaisums).
Achondroplasia ir autosomāli dominējošs ģenētisks traucējums.
Aarskoga sindroms ir ļoti reta iedzimta slimība, kurai raksturīgas galvas un sejas anomālijas (galvaskausa) zona, mazs vai vidēji īss augums, skeleta malformācijas un / vai dzimumorgānu anomālijas. Sejas vaibsti var ietvert plati izvietotas acis (acu hipertelorisms), noliektas uz leju plakstiņu krokas (acu spraugas), mazs deguns ar nāsīm un / vai augšžokļa nepietiekama attīstība (augšējā hipoplāzija) žoklis).
Skeleta anomālijas var ietvert neparasti īsus kāju pirkstus (brachydactyly), piekto pirkstu pastāvīgu fiksāciju saliektā stāvoklī (klinodactyly) un / vai neparasti platus īkšķus. Dzimumorgānu anomālijas var ietvert neparastu ādas kroku, kas stiepjas ap dzimumlocekļa pamatni, un / vai vienas vai abu sēklinieku nespēju nolaisties sēkliniekos (kriptorichisms). Dažiem skartajiem bērniem ir viegla garīga atpalicība.
Aarskoga sindroms ir ar X saistīta ģenētiska slimība.
Omodisplāzija Ir reta iedzimta kaulu slimība, ko var raksturot ar dažu roku garo kaulu (piemēram, augšdelma kaula) un kāju (t.i., augšstilba) patoloģisku īsumu kauli), īss augums un raksturīgas sejas vaibsti, ieskaitot nospiestu deguna tiltu, plašu deguna pamatni un / vai neparasti garu vertikālu gropi (gropi) augšdaļas centrā lūpas.
Turklāt skartajiem bērniem var būt patoloģiska divu apakšdelma kaulu (radiāla) atdalīšanās diastāze), radiālās galvas dislokācija un / vai noapaļotas kaulu masas (kondila) nepietiekama attīstība (hipoplāzija) beigās plecs.
Omodisplāziju var mantot kā autosomāli recesīvu vai autosomāli dominējošu ģenētisku stāvokli.