Липоидоза (лизозомална болест складиштења липида): шта је то, примери
Пажња! Информације су само у информативне сврхе и нису намењене за дијагнозу и преписивање лечења. Увек се консултујте са својим лекаром специјалистом!
Садржај
- Шта су лизозомалне болести складиштења?
- Шта су липиди?
- Како се липоидоза наслеђује?
- Врсте и симптоми лизозомалних болести складиштења
Шта су лизозомалне болести складиштења?
Лизозомалне болести складиштења (липоидоза), су група наследних метаболичких болести код којих се штетне количине масних материја (липида) акумулирају у различитим ћелијама и ткивима организма.
Људи са овим поремећајима или не производе довољно једног од ензима потребних за разградњу (метаболизацију) липида, или производе ензиме који не функционишу како треба.
Временом, ово прекомерно накупљање масти може довести до трајног оштећења ћелија и ткива, посебно у мозгу, периферном нервном систему (нерви од кичмене мождине до остатка тело), јетра, слезина и коштане сржи.
Шта су липиди?
Липиди су супстанце сличне мастима које су важни делови мембрана унутар и између ћелија и у мијелинском омотачу, који покрива и штити нерве. Липиди укључују уља, масне киселине, воскове, стероиде (као што су холестерол и естроген) и друга сродна једињења.
Ове масне супстанце се природно складиште у ћелијама, органима и ткивима тела. Сићушна тела унутар ћелија, названа лизозоми, редовно претварају или метаболишу липиде и протеине у мање компоненте како би обезбедили енергију телу. Поремећаји у којима интрацелуларни материјал, који се не може метаболисати, остаје у лизозомима, називају се болестима лизозома.
Поред болести повезаних са акумулацијом липида, друге лизозомске болести повезане са акумулацијом укључују муколипидозе, у којима се акумулирају ћелије и ткива прекомерна количина липида са молекулима шећера везаних за њих, као и мукополисахаридозе, у којима се акумулира превелика количина великог, сложеног шећера молекуле.
Како се наслеђују липоидоза?
Лизозомалне болести складиштења су наслеђене од једног или оба родитеља који носе дефектан ген који регулише специфични ензим који метаболизира липиде у класи ћелија у телу. Могу се наследити на два начина:
- Аутосомно рецесивно наслеђе јавља се када оба родитеља носе и преносе копију дефектног гена, али ниједан родитељ није погођен поремећајем. Свако дете рођено од ових родитеља има 25 одсто шансе да наследи обе копије дефектног гена, 50 одсто вероватноћа да ће бити носилац сличан својим родитељима и 25 одсто шансе да не наследи ниједну копију неисправног ген. Деца оба пола могу се наследити на аутозомно рецесиван начин.
- Кс-везано (или полно) рецесивно наслеђе настаје када мајка носи захваћени ген на Кс хромозому. Кс и И хромозоми су укључени у одређивање пола. Жене имају два Кс хромозома, док мушкарци имају један Кс хромозом и један И хромозом. Синови носилаца имају 50 посто шансе да наследе и утичу на поремећај, јер синови добијају један Кс хромозом од мајке и један И хромозом од оца. Ћерке имају 50 процената шансе да наследе захваћени Кс хромозом од своје мајке и да су носиоци или благо погођени. Оболели мушкарци не преносе болест на своје синове, али ће њихове ћерке бити носиоци и носиоци болести.
Врсте и симптоми лизозомалних болести складиштења
Гауцхерова болест - најчешћа од лизозомских болести. Болест је узрокована недостатком ензима глукоцереброзидазе, што доводи до акумулације глукоцереброзида у многим ткивима. Масни материјал се може акумулирати у мозгу, слезини, јетри, бубрезима, плућима и коштаној сржи.
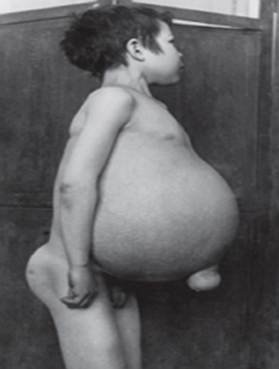
Симптоми могу укључивати оштећење мозга, повећање слезине и јетра, оштећена функција јетре, поремећаји скелета и оштећења костију која могу изазвати бол и преломе, отицање лимфних чворова и (понекад) суседних зглобова, надимање, браонкасти тон коже, анемија, низак број тромбоцита и жуте мрље на очима.
Најозбиљније погођени појединци могу такође бити подложнији инфекцијама. Болест подједнако погађа мушкарце и жене.
Гауцхерова болест има три општа клиничка подтипа:
- Тип 1 (Или не неуронопатски тип) је најчешћи облик болести у Сједињеним Државама и Европи. Мозак није захваћен, али може доћи до оштећења плућа и, у ретким случајевима, бубрега. Симптоми могу почети рано у животу или у одраслом добу и укључују повећану јетру и озбиљно повећање слезине, што може довести до руптуре и додатних компликација. Слабост скелета и болести костију могу бити опсежне. Људи из ове групе обично лако добију модрице због ниског броја тромбоцита у крви. Они такође могу искусити умор због анемије. У зависности од појаве и тежине болести, људи са тип 1 може да доживи пунолетство. Многи оболели имају благу болест или можда не показују никакве симптоме. Мада 1тип Гошеова болест је уобичајена међу људима ашкенаског јеврејског наслеђа и може утицати на људе свих етничких група.
- Тип 2 (или акутна неуропатија у детињству Гауцхерова болест) обично почиње у року од 3 месеца од рођења. Симптоми укључују екстензивно и прогресивно оштећење мозга, спастичност, нападе, укочене удове, увећану јетру (хепатомегалија јетре) и слезине (спленомегалија), абнормални покрети очију и слаба способност сисања и гутања. Погођена деца обично умиру пре 2 године.
- Тип 3 (хронични неуронопатски облик) може почети у било ком тренутку током детињства или чак у одраслом добу. Одликује се споро прогресивним, али мање тешким неуролошким симптомима у поређењу са акутном Гауцхеровом болешћу 2 врсте. Главни симптоми укључују поремећаје покрета очију, когнитивне дефиците, лошу координацију, нападе, повећање слезине и/или јетре, поремећаји скелета, поремећаји крви укључујући анемију и проблеми са дисање. Скоро сви са Гауцхеровом болешћу 3 врсте, који прима терапију замене ензима доживеће пунолетство.
За пацијенте 1. и 3. врсте Ензимска супституциона терапија, која се даје интравенозно сваке две недеље, може драматично смањити величину јетре и слезине, смањити абнормалности скелета и преокренути друге манифестације. Успешна трансплантација коштане сржи лечи неуролошке манифестације болести. Међутим, ова процедура носи значајне ризике и ретко се ради код особа са Гауцхеровом болешћу.
У ретким случајевима може бити потребна операција за уклањање целе или дела слезине (ако особа има веома низак број тромбоцита или када увећани орган у великој мери утиче на удобност особе). Трансфузија крви може бити од користи неким људима са анемијом. Другима ће можда бити потребна операција замене зглобова да би се побољшала мобилност и квалитет живота. Тренутно не постоји ефикасан третман за оштећење мозга које се може јавити код људи са типом 2 и 3 Гауцхерове болести.
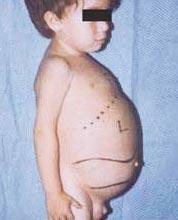
Ниеманн-Пицкова болест је група аутозомно рецесивних поремећаја узрокованих акумулацијом масти и холестерола у ћелијама јетре, слезине, коштане сржи, плућа и, у неким случајевима, мозга.
Неуролошке компликације могу укључивати атаксију (недостатак координације мишића који може утицати на ходање, писање и јело, између осталих функција), парализа ока, дегенерација мозга, проблеми са учењем, спастичност, отежано храњење и гутање, нејасно говор, губитак мишићног тонуса, преосетљивост на додир и извесна замућеност рожњаче због прекомерне акумулације материјала. Код 50% оболелих развија се карактеристичан ореол трешње црвене боје око центра мрежњаче, који лекар може да види посебним инструментом.
Ниеманн-Пицкова болест је класификована у три општа подтипа:
- Укуцате, најтежи облик, почиње у раном детињству. Бебе изгледају нормално при рођењу, али до 6 месеци развија се дубоко оштећење мозга, увећана јетра и слезина, и отечени лимфни чворови и чворови испод коже (ксантоми). Слезина може постати 10 пута већа од нормалне величине и може пукнути, узрокујући крварење. Ова деца прогресивно постају слабија, губе моторичку функцију, могу постати анемична и склона су поновним инфекцијама. Ретко живе дуже од 18 месеци. Овај облик болести је најчешћи у јеврејским породицама.
- Тип Б(или јувенилни почетак) обично не утиче на мозак, али већина деце се развија атаксија, оштећење нерава који излазе из кичмене мождине (периферна неуропатија), и плућне потешкоће које напредују са годинама. Повећање јетре и слезине је типично за адолесценте. Људи са тип Б могу да живе релативно дуго, али многима је потребна доживотна терапија кисеоником због оштећења плућа. Ниеманн-Пицк типови А и Б су резултат акумулације масне супстанце зване сфингомијелин због недостатка ензима званог сфингомијелиназа.
- Тип Ц може се појавити рано у животу или развити током адолесценције или чак одраслог доба. Ниеманн-Пицкова болесттип Ц није узрокован недостатком сфлингомијелиназе, већ недостатком НПЦ1 или НПЦ2 протеина. Као резултат тога, различити липиди и посебно холестерол се акумулирају у нервним ћелијама и узрокују њихов квар. Захваћеност мозга може бити опсежна, што резултира немогућношћу гледања горе-доле, потешкоћама у ходању и гутању, прогресивним губитком слуха и прогресивним деменција. Људи са тип Ц имају само благо повећање слезине и јетре. Очекивано трајање живота људи са тип Ц знатно варира. Неки људи умиру у детињству, док други који су мање погођени могу такође живети у одраслом добу.
Тренутно не постоји лек за Ниеманн-Пицкову болест. Лечење је потпорно. Деца обично умиру од инфекције или прогресивног неуролошког губитка. Било је случајева трансплантације коштане сржи код неколико пацијената са тип Б са мешовитим резултатима.
Фабријева болесттакође познат као недостатак алфа-галактозидазе-А, изазива накупљање масне материје у аутономном нервном систему (оном делу нервног система који контролише невољне функције као што су дисање и рад срца), у очима, бубрезима и кардиоваскуларним система.
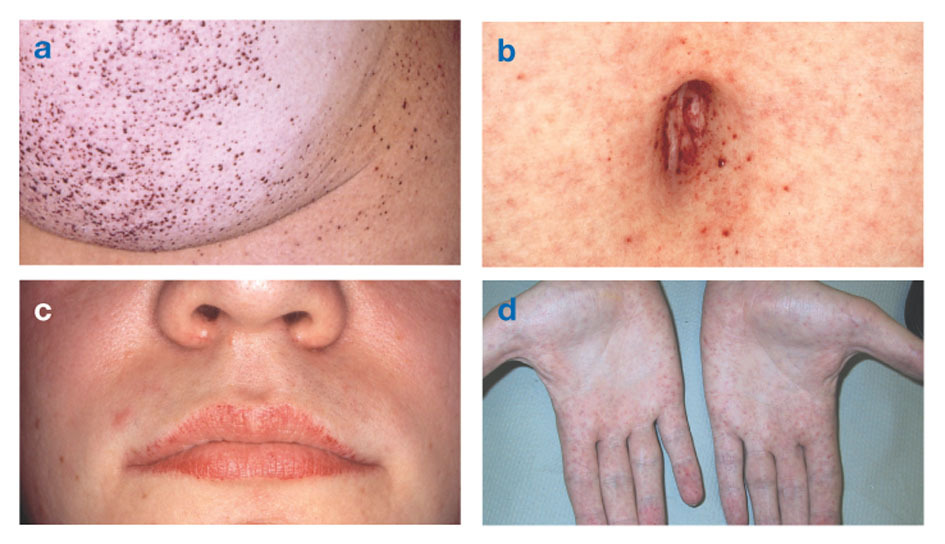
Фабријева болест је једина болест липида везана за Кс. Болест углавном погађа мушкарце, мада се блажи и варијабилнији облик јавља код жена. Ретко, погођене жене имају тешке симптоме сличне онима који се виде код мушкараца са овим поремећајем. Почетак симптома обично се јавља током детињства или адолесценције.
Неуролошки знаци укључују пекуће болове у рукама и ногама који се погоршавају по врућем времену или после вежбање, као и накупљање вишка материјала у провидним слојевима рожњаче (што доводи до замућења, али без промене вида). Акумулација масти у зидовима крвних судова може пореметити циркулацију, доводећи особу у опасност удар или срчани напад. Остали симптоми укључују увећано срце, прогресивно бубрежна инсуфицијенцијашто доводи до завршног стадијума бубрежне болести, гастроинтестиналних проблема, смањеног знојења и грознице.
Ангиокератоми (мале, неканцерозне, црвенкасто-љубичасте подигнуте мрље на кожи) могу се развити у доњем делу трупа и постати чешћи са годинама.
Људи са Фабријевом болешћу често умиру прерано од компликација од отказивање срца, отказивање бубрега или мождани удар. Лекови као што су фенитоин и карбамазепин се често прописују за лечење бола који прати Фабријеву болест, али не и за његово лечење. Метоклопрамид или Липисорб (додатак исхрани) могу ублажити гастроинтестиналне сметње често се јавља код људи са Фабријевом болешћу, а неким људима може бити потребна трансплантација бубрега или дијализа. Замена ензима може смањити складиштење, ублажити бол и сачувати функцију органа код неких људи са Фабријевом болешћу.
Фарберова болест такође познат као Фарберова липогрануломатоза, описује групу ретких аутозомно рецесивних поремећаја који изазивају накупљање масне материје у зглобовима, ткивима и централном нервном систему. Погађа и мушкарце и жене. Болест обично почиње у раном детињству, али се може јавити и касније у животу.
Развијају се деца са класичном Фарберовом болешћу неуролошки симптоми, који могу укључивати повећану поспаност и летаргију и проблеме са гутање. Такође могу бити погођени јетра, срце и бубрези. Остали симптоми могу укључивати контрактуре зглобова (хронично скраћивање мишића или тетива око зглобова), повраћање, артритис, увећање лимфни чворови, упала зглобова, промуклост и квржице испод коже које се згушњавају око зглобова како напредују болести.
Погођеним особама са потешкоћама у дисању ће можда бити потребно убацити цев за дисање. Већина деце са овим стањем умире до 2 године, обично од болести плућа. Код једног од најтежих облика болести, увећана јетра и слезина могу се дијагностиковати убрзо након рођења. Бебе рођене са овим обликом болести обично умиру у року од 6 месеци.
Фарберова болест је узрокована недостатком ензима званог церамидаза. Тренутно не постоји специфичан третман за Фарберову болест. Кортикостероиди се могу прописати за ублажавање болова. Трансплантација коштане сржи може смањити грануломе (мале масе запаљеног ткива) код људи са благим упалом или код пацијената без компликација на плућном или нервном систему. Код старијих људи, грануломи се могу смањити или хируршки уклонити.
Ганглиозидоза састоји се од две различите групе генетских болести. Оба су аутозомно рецесивна и подједнако утичу на мушкарце и жене.
ГМ1 ганглиозидоза.
ГМ1 ганглиозидоза је узрокована недостатком ензима бета-галактозидазе, што доводи до абнормалне акумулације кисели липидни материјали, посебно у нервним ћелијама у централном и периферном нервном систему система. ГМ1 ганглиозидоза има три клиничке манифестације:
- ГМ1 (најтежи подтип, који се јавља убрзо након рођења) може укључивати неуродегенерацију, нападе, увећану јетру и слезину, грубост црта лица, неправилности скелета, укоченост зглобова, надимање, слабост мишића, претерани одговор на препад и проблеми са ход. Отприлике половина оболелих развије мрље боје трешње у оку. Деца могу бити глува и слепа до прве године и често умру до 3 године од срчаних компликација или упала плућа.
- Касно дете ГМ1 ганглиозидоза обично почиње између 1 и 3 године. Неуролошки симптоми укључују атаксију, нападе, деменцију и потешкоће у говору.
- ГМ1 ганглиозидоза развија се у доби од 3 до 30 година. Симптоми укључују смањену мишићну масу (атрофију мишића), неуролошке компликације које су мање тешке и напредују спорије од других облика поремећаји, непрозирност рожњаче код неких људи и трајне контракције мишића које узрокују увртање и понављајуће покрете или абнормалне положаје (дистонија). Ангиокератоми се могу развити у доњем делу трупа. Величина јетре и слезине је нормална код већине оболелих људи.
ГМ2 ганглиозидоза.
ГМ2 ганглиозидоза такође узрокује да тело акумулира вишак киселих масних супстанци у ткивима и ћелијама, првенствено у нервним ћелијама. Ови поремећаји су резултат недостатка ензима бета-хексосаминидазе. ГМ2 поремећаји укључују:
- Таи-Сацхсова болест (такође познат као ГМ2 ганглиозидоза варијанта Б) и њени варијантни облици узроковани су недостатком ензима хексосаминидазе А. Погођена деца се нормално развијају током првих неколико месеци живота. Симптоми почињу са 6 месеци и укључују прогресивни губитак менталних способности, деменцију, смањен контакт очима, повећан запањујући одговор на буку, прогресивни губитак слуха који доводи до глувоће, отежано гутање, слепило, трешње црвене мрље на мрежњачи и неке парализа. Напади могу почети у другој години живота детета. Бебе могу на крају постати зависне од сонде за храњење и често умру до 4 године од понављајућих инфекција. Нажалост, не постоји посебан третман. Антиконвулзанти могу у почетку да контролишу нападе. Остали третмани подршке укључују правилну исхрану и хидратацију, као и методе за одржавање дисајних путева отвореним. Редак облик поремећаја тзв касни почетак Таи-Сацхсове болести, јавља се код људи између 20 и 30 година и карактерише је нестабилан ход и прогресивно неуролошко погоршање.
- Сандхоффова болест (Опција АБ) је тежак облик Таи-Сацхсове болести. Почетак се обично јавља у доби од 6 месеци и није ограничен на било коју етничку групу. Неуролошки знаци могу укључивати прогресивно погоршање централног нервног система, мотор слабост, рано слепило, тешка реакција уплашености на звук, спастичност, шок или трзање мишићи (миоклонус), епилепсија, абнормално увећана глава (макроцефалија) и црвене мрље у оку. Остали симптоми могу укључивати честе респираторне инфекције, шум на срцу, црте лица попут лутке и повећање јетре и слезине. Не постоји специфичан третман за Сандхоффову болест. Као и код Таи-Сацхсове болести, помоћна нега укључује одржавање дисајних путева отвореним у сваком тренутку, као и правилну исхрану и хидратацију. Антиконвулзанти могу у почетку да контролишу епилепсију (нападе).
Краббеова болест(такође познат као леукодистрофија глобуларних ћелија и галактозилцерамид липидоза) је аутозомно рецесивна болест узрокована недостатком ензима галактоцереброзидазе. Болест најчешће погађа одојчад са почетком пре 6 месеци старости, али се може јавити током адолесценције или одраслог доба.
Акумулација несварених масти утиче на раст заштитног омотача нерва (мијелинске овојнице) и изазива озбиљно оштећење менталних и моторичких способности. Остали симптоми укључују слабост мишића, смањену способност мишића да се истегну (хипотонија), напетост мишића (спастичност), изненадни шок или трзање удова (миоклонични напади), раздражљивост, необјашњива грозница, глувоћа, слепило, парализа и отежано гутање. Може доћи и до дуготрајног губитка тежине.
Болест се може дијагностиковати испитивањем ензима и идентификовањем карактеристичних група ћелија у глобоидна тела у белој материји мозга, демијелинизација нерава и дегенерација и уништавање можданих ћелија. Код одојчади, болест је обично фатална пре 2 године живота. Особе са узнапредовалим обликом болести имају блажи ток болести и живе знатно дуже.
Није развијен специфичан третман за Краббеову болест, иако рана трансплантација коштане сржи може помоћи неким људима. Особе са узнапредовалим обликом болести имају блажи ток болести и живе знатно дуже.
Метахроматска леукодистрофија, или МЛД, је група поремећаја обележених акумулацијом масних материја у белој материји централног нервног система и периферним нервима и донекле у бубрезима. Слично Краббеовој болести, МЛД утиче на мијелин, који покрива и штити нерве. Овај аутозомно рецесивни поремећај је узрокован недостатком ензима арилсулфатазе А. Овај поремећај погађа и мушкарце и жене.
Метахроматска леукодистрофија има три карактеристична облика: касно дојенчад, јувенилни и одрасли.
- Лате Инфант МЛД обично почиње између 12 и 20 месеци након рођења. Деца у почетку могу изгледати нормално, али имају потешкоће у ходању и склоност падању, праћена повременим болом у рукама и ногама. прогресивни губитак вида који доводи до слепила, кашњења у развоју и губитка претходно стечених прекретница, отежаног гутања, нападаја и деменције до 2 године. Деца такође развијају прогресивно трошење мишића и слабост и на крају губе способност ходања. Већина деце са овим обликом поремећаја умире до 5. године.
- Јувениле МЛД обично почиње између 3 и 10 година. Симптоми укључују смањен школски успех, ментално оштећење, атаксију, нападе и деменцију. Симптоми напредују, а смрт наступа 10-20 година након појаве болести.
- Форма за одрасле. Симптоми одрасли почиње након 16. године и може укључивати атаксију, нападе, абнормални тремор удова (дрхтање), смањену концентрацију, депресија, менталних поремећаја и деменције. Смрт обично наступа 6-14 година након појаве симптома.
МЛД се не може излечити. Лечење је симптоматско и подржавајуће. Трансплантација коштане сржи може у неким случајевима одложити напредовање болести. Значајан напредак је постигнут са генском терапијом на животињским моделима са МЛД и у клиничким испитивањима.
Волманова болесттакође познат као недостатак лизозомалне киселе липазеје озбиљан поремећај складиштења липида који је обично фаталан у доби од 1 године. Овај аутозомно рецесивни поремећај карактерише акумулација естара холестерола (обично транспортни облик холестерола) и триглицериди (хемијски облик у којем масти постоје у телу), који се могу значајно акумулирати и узроковати оштећење ћелија и тканине.
И мушкарци и жене пате од овог поремећаја. Деца су нормална и активна при рођењу, али брзо развијају прогресивно ментално оштећење, увећану јетру и знатно увећану слезину, надимање, гастроинтестиналне проблеме. жутица, анемија, повраћање и депозити калцијума у надбубрежне жлездеузрокујући њихово стврдњавање.
Друга врста недостатак лизозомалне киселе липазе је болест складиштења естара холестерола. Ова изузетно ретка болест настаје као резултат складиштења естара холестерола и триглицериди у крвним ћелијама, лимфа и лимфоидно ткиво. Код деце у одраслом добу, јетра се повећава, што доводи до цирозе и хронична инсуфицијенција јетре. Деца могу имати и наслаге калцијума у надбубрежним жлездама, а жутица се може развити у каснијим стадијумима болести.
Тренутно се питање замене ензима активно проучава како у Волмановој болести, тако иу акумулацији естра холестерола.
Пажња! Информације су само у информативне сврхе и нису намењене за дијагнозу и преписивање лечења. Увек се консултујте са својим лекаром специјалистом!