Thalassemie: wat is deze ziekte, symptomen, analyses, typen (classificatie)
Inhoud
- Wat is thalassemie?
- Thalassemie classificatie
- Alfa-thalassemie
- Bèta-thalassemie
- Welke onderzoeken, analyses zijn nodig?
- Thalassemie behandeling
- Gerelateerde video's
Wat is thalassemie?
Thalassemie (bloedarmoede van Cooley) Is een groep erfelijke aandoeningen die de hoeveelheid hemoglobine die door een persoon wordt geproduceerd beïnvloeden. Hemoglobine behoort tot een familie van verbindingen die bestaat uit heem (een ijzercomplex) en verschillende globines (eiwitketens die het heemcomplex omringen).
Hemoglobine (Hb) moleculen komen voor in alle rode bloedcellen en zijn de oorzaak van hun rode kleur. Ze binden zuurstof in de longen, voeren het door de bloedbaan en geven het af aan lichaamsweefsels. De verschillende soorten hemoglobine worden ingedeeld naar het type eiwitketens dat ze bevatten.
Normale volwassen hemoglobines zijn onder meer:
- Hemoglobine A (maakt ongeveer 95% - 98% uit van het volwassen Hb). HbA bevat twee alfa (α) eiwitketens en twee bèta (ß) ketens.
- HbA2 (maakt ongeveer 2% - 3,5% uit van het volwassen Hb) heeft twee alfa (α) en twee delta (δ) ketens.
- HbF (tot 2%). Het is de belangrijkste hemoglobine die de foetus tijdens de zwangerschap aanmaakt. De productie daalt meestal binnen een jaar na de geboorte tot een laag niveau. HbF heeft twee alfa (α) en twee gamma (γ) ketens.
Mutaties in de genen die coderen voor globineketens kunnen verstoringen in de productie van hemoglobine veroorzaken. Er zijn 4 genen die coderen voor de alfaglobineketen en 2 genen die coderen voor de bètaglobineketen.
Erfelijke aandoeningen van de hemoglobineproductie vallen in twee categorieën:
- Thalassemie: verminderde productie van normaal hemoglobine
- Hemoglobinopathie: de productie van een abnormaal hemoglobinemolecuul
Thalassemieën zijn een groep aandoeningen waarbij mutaties in een of meer van de alfa- of bètaglobinegenen een afname van de geproduceerde hoeveelheid HbA veroorzaken. Dit leidt tot een verlaging van het HbA-niveau, een relatieve toename van de hoeveelheid kleine hemoglobines HbA2 en HbF en mogelijk de detectie van ongebruikelijke soorten hemoglobine.
 Genetica (klikbaar)
Genetica (klikbaar)Thalassemieën worden meestal geclassificeerd volgens het type globineketen, waarvan de synthese is verminderd.
Thalassemie classificatie
Alfa-thalassemie

Alfa-thalassemie is het gevolg van een deletie of mutatie in een of meer kopieën van het 4-alphaglobine-gen. Hoe meer genen worden aangetast, hoe minder alfaglobine wordt geproduceerd. De vier verschillende soorten alfa-thalassemie omvatten:
- Alfa-thalassemie-eigenschap (1 aangetast gen). De stille drager zal normale hemoglobinewaarden en rode bloedcellen hebben die normaal zijn of een licht verlaagde MCH (hypochromie) vertonen. Dragers kunnen het aangetaste gen doorgeven aan hun nakomelingen. Vaak worden deze mensen pas geïdentificeerd na de geboorte van een kind met HbH-ziekte of een teken van alfa-thalassemie.
- Alfa-thalassemie eigenschap (2 aangetaste genen). Patiënten met een teken van alfa-thalassemie hebben kleinere (microcytische), bleke (hypochrome) rode bloedcellen en milde chronische bloedarmoede, maar ze ervaren meestal geen symptomen. het Bloedarmoededie niet reageert op ijzersupplementen. Diagnose van het teken van alfa-thalassemie vindt meestal plaats door andere oorzaken van microcytaire anemie uit te sluiten. Bevestigend testen door middel van DNA-analyse is een belangrijk onderdeel van het diagnoseproces van alfa-thalassemie. omdat op elk chromosoom er zijn twee kopieën (HbA1 en HbA2) van het alfaketengen. Als een persoon beide exemplaren van één chromosoom heeft verloren, heeft dit verschillende gevolgen voor de overdracht van meer ernstige ziekte voor hun kinderen dan wanneer ze één exemplaar van elk van twee verschillende chromosomen. Zie het gedeelte over DNA-analyse hieronder.
- Hemoglobine H-ziekte (3 aangetaste genen). Onder deze voorwaarde veroorzaakt een grote afname van de hoeveelheid geproduceerde alfaglobineketens een overmaat bètaketens, die vervolgens aggregeren tot bèta4-tetrameren (groepen van 4 bètaketens), bekend als: hemoglobine H. HbH-ziekte kan matige tot ernstige bloedarmoede en splenomegalie (verhoogde milt). Het klinische beeld geassocieerd met HbH-ziekte is zeer divers. Bij sommige mensen is het asymptomatisch, terwijl anderen ernstige bloedarmoede hebben. De ziekte van hemoglobine H komt het meest voor bij mensen van zuidoostelijke of oostelijke mediterrane afkomst.
- Alfa-thalassemie major (ook wel foetale hydrops genoemd, 4 aangetaste genen). Dit is de meest ernstige vorm van alfa-thalassemie. Onder deze omstandigheden wordt alfaglobine niet geproduceerd, daarom wordt er ook geen HbA of HbF geproduceerd. Foetussen die worden aangetast door alfa-thalassemie worden bloedarmoede in het begin van de zwangerschap. Ze worden oedemateus (houden vocht vast) en hebben vaak vergrote harten en lever. Deze diagnose wordt vaak gesteld in de laatste maanden van de zwangerschap wanneer een echografie van de foetus foetaal oedeem aangeeft. In ongeveer 80% van de gevallen ontwikkelt de moeder toxemie en kan ernstige postpartumbloeding ontstaan. Baby's met alfa-thalassemie major zijn meestal prematuur, doodgeboren of overlijden kort na de geboorte.
Lees ook:Oorzaken van verhoogde hemoglobine in het bloed bij mannen, zoals gemanifesteerd, klachten, behandeling
Mensen met alfa-thalassemie kunnen door onoplettende artsen verkeerd worden gediagnosticeerd als ijzertekort, aangezien ijzertekort ook leidt tot het verschijnen van kleine bleke (microcytische hypochrome) erytrocyten.
Belangrijkdat ijzerbehandeling bij thalassemiepatiënten alleen wordt voorgeschreven wanneer specifieke ijzertesten (ferritine, serumijzer, TIBC, transferrine) ijzertekort hebben bevestigd. Dit is vooral belangrijk bij alfa-thalassemie waar een kleine kans bestaat op het ontwikkelen van een gevaarlijke ijzerstapeling.
Alfa-thalassemie komt het meest voor bij mensen met een etnische achtergrond uit Zuidoost-Azië, Zuid-China, het Midden-Oosten, India, Afrika en de Middellandse Zee.
Bèta-thalassemie

Bèta-thalassemie treedt op als gevolg van mutaties in een of beide bètaglobinegenen. Er zijn tussen de 100 en 200 mutaties geïdentificeerd, maar slechts ongeveer 20 komen vaak voor.
De ernst van bèta-thalassemie-anemie hangt af van welke mutaties aanwezig zijn en of ze verminderen de productie van bètaglobine (de zogenaamde bèta + thalassemie) of volledig elimineren (de zogenaamde bèta-thalassemie). De verschillende soorten bèta-thalassemie zijn als volgt:
- Beta-thalassemie met karakteristieke. Een persoon met deze ziekte heeft één normaal gen en één met een mutatie. Ze ervaren meestal geen andere gezondheidsproblemen dan microcytose (kleine rode bloedcellen) en mogelijk milde bloedarmoede die niet reageert op ijzersupplementen. Deze genmutatie kan worden doorgegeven aan de kinderen van een individu.
- Thalassemie intermedia. In deze toestand heeft de zieke persoon twee abnormale genen, maar ze produceren nog steeds wat bètaglobine. De ernst van de bloedarmoede die optreedt en gezondheidsproblemen zijn afhankelijk van de aanwezige mutaties. De scheidslijn tussen thalassemie intermedia en thalassemie major is de mate van bloedarmoede en het aantal en de frequentie van bloedtransfusies die nodig zijn om het te behandelen. Degenen met thalassemie intermedia hebben soms transfusies nodig, maar niet op regelmatige basis.
- Thalassemie major. Dit is de meest ernstige vorm van bèta-thalassemie. De patiënt heeft twee abnormale genen die ofwel een ernstige afname ofwel een volledig gebrek aan bètaglobineproductie veroorzaken, waardoor de productie van aanzienlijke hoeveelheden HbA wordt voorkomen. Deze aandoening verschijnt meestal bij een baby na de leeftijd van 3 maanden en veroorzaakt levensbedreigende bloedarmoede. Dit Bloedarmoede vereist gedurende het hele leven regelmatige bloedtransfusies en aanzienlijke voortdurende medische aandacht. Na verloop van tijd leiden deze frequente transfusies tot een teveel aan ijzer in het lichaam. Indien onbehandeld, kan dit overtollige ijzer worden afgezet in de lever, het hart en andere organen en kan dit leiden tot vroegtijdig overlijden door orgaanfalen.
Lees ook:De milt: wat is verantwoordelijk voor, waar te zijn, hoe het pijn doet, symptomen en behandeling
Andere vormen van thalassemie treden op wanneer het bèta-thalassemie-gen wordt geërfd in combinatie met het hemoglobinevariant-gen. De belangrijkste hiervan zijn:
- HbE - bèta-thalassemie. HbE is een van de meest voorkomende hemoglobinevarianten en wordt voornamelijk aangetroffen bij mensen van Zuidoost-Aziatische afkomst. Als een persoon één HbE-gen en één bèta-thalassemie-gen erft, veroorzaakt de combinatie HbE bèta-thalassemie, die matig ernstige bloedarmoede veroorzaakt, vergelijkbaar met bèta-thalassemie intermedia.
- HbS - bèta-thalassemie of sikkelcel - bèta-thalassemie. HbS is een van de bekendste hemoglobinevarianten. Overerving van één HbS-gen en één bèta-thalassemiegen resulteert in HbS bèta-thalassemie. De ernst van de aandoening hangt af van de hoeveelheid bètaglobine die door het bèta-gen wordt geproduceerd. Als bètaglobine niet wordt geproduceerd, is het klinische beeld bijna identiek aan sikkelcelziekte.
Welke onderzoeken, analyses zijn nodig?
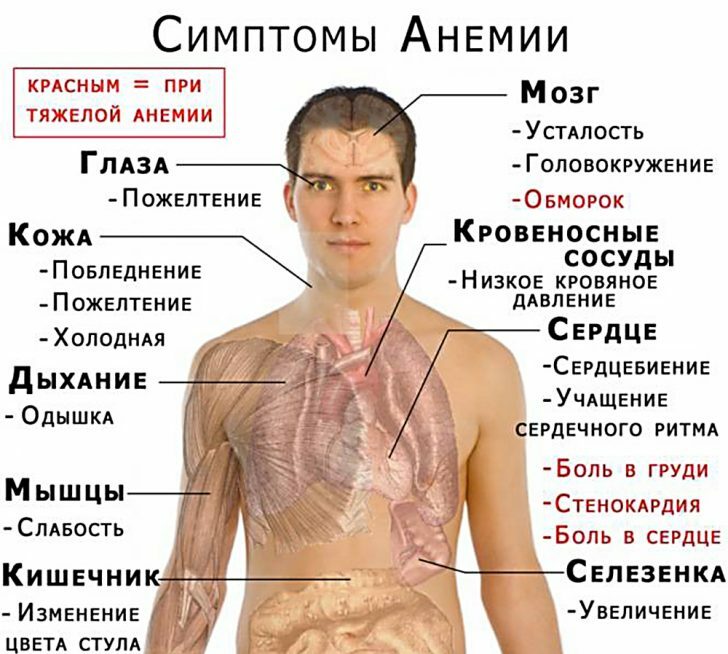
Algemeen klinisch bloedonderzoek (CBC). KLA is een analyse van cellen en vocht in het bloed. Het KLA zal de arts onder meer vertellen hoeveel rode bloedcellen er in het bloed zitten, hoeveel hemoglobine ze bevatten en helpen de grootte en vorm van de aanwezige rode bloedcellen te meten. Deze variabelen worden erytrocytenindexen genoemd en omvatten MCH (gemiddeld corpusculair hemoglobine) en MCV (gemiddeld corpusculair volume) als metingen van het hemoglobinegehalte en de grootte van rode bloedcellen.
Lage MCH of MCV zijn vaak de eerste tekenen van thalassemie. Als de MCH- of MCV-spiegels laag zijn en ijzertekort is uitgesloten, kan de persoon drager zijn van thalassemie. Bij ijzertekort is thalassemie moeilijk uit te sluiten.
Microscopisch onderzoek van een bloeduitstrijkje. Bij deze test wordt een dun microscopisch laagje bloed op een glasplaatje onder een microscoop onderzocht. Het aantal en type witte bloedcellen, rode bloedcellen en bloedplaatjes kunnen handmatig worden geteld en beoordeeld als ze normaal en volwassen zijn. Verschillende aandoeningen beïnvloeden de normale productie van bloedcellen. Bij thalassemie zijn rode bloedcellen vaak microcytisch (klein) met een lage MCV. Ze kunnen ook:
- Hypochroom zijn (bleek - geeft aan) lage hemoglobine, dan normaal).
- Variëren in grootte (anisocytose) en vorm (poikilocytose).
- Een foetus zijn (niet normaal in een volwassen rode bloedcel).
- Wees vervormd, zie eruit als appels onder de microscoop.
IJzeranalyse. De test kan het volgende omvatten: IJzer, ferritine, UIBC, TIBC en transferrinepercentageverzadiging. Deze tests meten verschillende aspecten van de opslag en het gebruik van ijzer in het lichaam. Ze helpen bepalen of ijzertekort de oorzaak en/of verergering van de bloedarmoede van de patiënt is. Een of meer patiënten met thalassemie kunnen ook worden gecontroleerd op de mate van ijzerstapeling.
Beoordeling van hemoglobinopathie (Hb). Deze test meet het type en de relatieve hoeveelheid hemoglobine die aanwezig is in rode bloedcellen. Hemoglobine A, bestaande uit alfa- en bètaglobine, is het belangrijkste normale type hemoglobine dat bij volwassenen wordt aangetroffen. Een hoger percentage HbA2 en/of HbF wordt meestal gezien wanneer bèta-thalassemie aanwezig is. HbH kan worden gezien bij alfa-thalassemie, maar alleen wanneer ten minste twee van de vier alfa-genen zijn verwijderd of gemuteerd.
Lees ook:Verhoogde eosinofielen in het bloed bij een volwassene
DNA-analyseDNA-analyse is in veel gevallen niet nodig, omdat uit de resultaten van bovenstaande tests een diagnose kan worden gesteld. DNA-testen worden het meest gebruikt in families met alfa-thalassemie. Deze test wordt gebruikt om deleties en mutaties in alfa- en minder vaak bètaglobine-producerende genen te bestuderen.
Familie onderzoek kan worden uitgevoerd om de status van de drager en de soorten mutaties in andere familieleden te beoordelen. DNA-onderzoek is een belangrijk hulpmiddel voor het stellen van een nauwkeurige diagnose van thalassemie. Vooral bij alfa-thalassemie is het belangrijk om te weten of een persoon met de eigenschap alfa-thalassemie twee gemuteerde genen op één chromosoom of één op elk chromosoom heeft.
Thalassemie behandeling
De meeste mensen met thalassemie hebben geen behandeling nodig. Alle mensen met de diagnose thalassemie die een gezin plannen, wordt sterk aangeraden om te zoeken naar genetisch adviesom de gevolgen voor hun nakomelingen te begrijpen. Is het vereist dat het laboratorium heeft een genetische studie uitgevoerd partner zodat de geneticus nauwkeurige informatie kan verstrekken over het risico van het doorgeven van de aangetaste genen aan hun kinderen en de ernst van thalassazemie of hemoglobinopathie die zich kan ontwikkelen.
Patiënten met hemoglobine H of bèta-thalassemie intermedia zullen hun hele leven verschillende gradaties van bloedarmoede ervaren. Ze kunnen een relatief normaal leven leiden, maar vereisen: regelmatige controle en soms misschien nodig bloedtransfusie.
Foliumzuursupplementen worden vaak voorgeschreven om bloedarmoede te bestrijden, maar ijzersupplementen moeten worden vermeden, tenzij ijzertekort wordt bevestigd door meer specifieke onderzoeken.
Veranderingen in het aantal rode bloedcellen worden vaak waargenomen wanneer een vrouw zwanger is, en dit kan soms leiden tot ernstige bloedarmoede. Het begrijpen van de impact van een thalassemiediagnose op de zwangerschap en de mogelijke gevolgen voor de gezondheid van het nageslacht van een paar is een van de belangrijkste redenen om een nauwkeurige diagnose van thalassemie te stellen.
Patiënten met bèta-thalassemie hebben hun hele leven gewoonlijk elke 3-4 weken een bloedtransfusie nodig. Deze transfusies helpen de hemoglobineconcentratie hoog genoeg te houden om het lichaam van zuurstof te voorzien en groeistoornissen en orgaanschade te voorkomen.
Frequente transfusies verhogen echter het ijzer tot toxische niveaus, wat leidt tot ijzerafzetting in de lever, het hart en andere organen.
Regelmatige ijzerchelatietherapie gebruikt om het ijzergehalte in het lichaam te verlagen.Dit omvat de toediening van een medicijn dat zich aan het ijzer bindt en helpt het via de urine uit het lichaam te spoelen. Splenectomie (operatie om de milt te verwijderen) komt ook vaak voor.
Baby's met alfa-thalassemie major worden meestal te vroeg geboren, doodgeboren of sterven kort na de geboorte. De behandeling is gericht op het identificeren van de aandoening en het beëindigen van de zwangerschap of het controleren op complicaties bij de moeder.
Experimentele behandelingen zoals foetale bloedtransfusies zijn in zeer weinig gevallen bij de geboorte succesvol geweest.