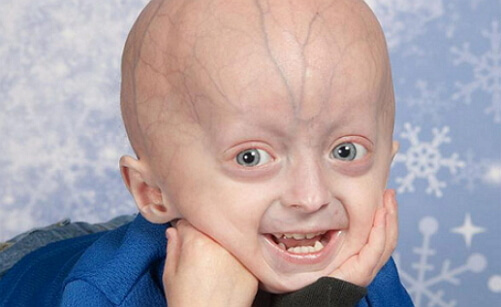
Nepopolna osteogeneza
 Nepravilna osteogeneza je prirojena bolezen kosti in posameznih struktur vezivnega tkiva. Drugo ime za nepopolno osteogenezo je bolezen krhkih kosti. Bolezen se prenaša z dedovanjem, t.j. Je genetsko določen.
Nepravilna osteogeneza je prirojena bolezen kosti in posameznih struktur vezivnega tkiva. Drugo ime za nepopolno osteogenezo je bolezen krhkih kosti. Bolezen se prenaša z dedovanjem, t.j. Je genetsko določen.
Neučinkovita osteogeneza pri moških in ženskah je incidenca 1 novorojenca na 12 000-15 000 otrok. Ta patologija je bila prvič omenjena v 17. stoletju. V nepopolni osteogenezi so moteni rast, kožne lezije, mišično tkivo, krvne žile, zobje, kite in sluh.
Kljub težavam, s katerimi se soočajo ljudje z neuspešno osteogenezo, večina bolnikov vodi celovit življenjski slog. Trenutno zdravljenje nepopolne osteogeneze postaja vse bolj priljubljeno. Kljub temu pa je še vedno nemogoče popolnoma zdraviti bolnika s tako boleznijo.
Nepravilna osteogeneza vzroka
Nepravilna osteogeneza se nanaša na heterogene dedne bolezni, ki vplivajo na vezivno tkivo. Za bolezen je značilna osteopenija in nekateri drugi klinični znaki osteogeneze.
Glavni vzrok tega razvoja patologije je dedna mutacija kolagenih genov. Redki je spontani pojav mutacij v genih tega proteina. Posledica genske mutacije je kršitev sinteze kolagena, zato so patologije pri nastanku kostnega in krvavega tkiva.
Nezadostna količina sintetiziranega kolagena( ki ni mutirana) je tudi eden od razlogov za nastanek nepopolne osteogeneze. V tem primeru bolezen poteka v bolj blagi obliki. Nepopolna osteogeneza te vrste je izražena v posameznih zlomih udov, po puberteti se število zlomov praviloma zmanjša.
Sodobna medicina razlikuje več vrst nepopolne osteogeneze ob upoštevanju radiografskih, kliničnih, kolagenskih proteogenih sprememb.
Nepravilne vrste osteogeneze:
Tip I - šibko izražena oblika, prevladujoča vrsta dedovanja. Nepopolna osteogeneza, za katero so značilne krhke kosti, prisotnost modre sklere.
Tip II - perinatalno-smrtonosno.
Tip III - deformacija okostja je progresivna.
Tip IV - prevladujoča vrsta dedovanja, deformacije okostja niso naglo naglašene, sklerje so normalne.
Mnogi verjamejo, da se nepopolna osteogeneza razvija v ozadju kvalitativnih in kvantitativnih kršitev sinteze kolagena tipa I.Za neuspešno osteogenezo tipa I je značilna nižja stopnja sinteze normalne kolagene. Tip II, podobno kot nepopolna osteogeneza tipa IV, je posledica zmanjšane skupne količine kolagena zaradi zmanjšane stabilnosti, čeprav postopek sinteze kolagena poteka brez motenj.
Nepravilni simptomi osteogeneze
Vsaka vrsta nepopolne osteogeneze ima značilno simptomatologijo.
Torej, vrsta neusklajene osteogeneze pri otrocih se kaže z osteoporozo, kot tudi pogostimi zlomi kosti. Po desetletni starosti se pojavnost zlomov zmanjša, vendar se po 40 letih znova povečuje. Opazimo modro sclero in zgodnji pojav senilnega platišča. Bolniki( približno 50% primerov) s to vrsto nepopolne osteoporoze z nizko rastjo, nekateri dentin ostanejo normalni, nekateri bolniki imajo dentin opal. Pri tipu I se pojavijo spremembe v aortiki v številnih primerih, primanjkuje mitralne motnje( v 20% primerov pride do prolapsa mitralnega ventila), krvavitve iz nosu.
Tip II neuspešne osteogeneze je značilen zaradi smrti ploda pred rojstvom ali zgodnje smrti novorojenčkov. Zlomi so večkratni in pogosti, zlahka nastanejo. Obstajajo tri skupine: skupina
A. Poškodbe glave, okončine v plodu so opazne med nosečnostjo zaradi krhkosti oblik vezivnega tkiva. Območje možganov lobanje se poveča, prsni koš pa se zmanjša, okončine so kratke in ukrivljene. Obstajajo hude primere kalcifikacije stene aorte in endokarda. Takšni otroci so rojeni zelo majhno rast( približno 25-30 cm).
Births pred terminom, z 20% takih prezgodnjih rojstev - mrtvorojenjem. Nekateri bolni otroci umirajo v zgodnjih dneh, nekateri živijo do četrtega tedna. Patološke spremembe je mogoče odkriti še pred rojstvom otroka s pomočjo rentgenske aparate: široka stegna, ima valovite robove, robove z rožnega venca, prsih kratko. Pri izvajanju medicinskega genetskega svetovanja pri poskusih obstaja možnost molekularne pomanjkljivosti v kombinaciji s heterozigotnostjo mutacij, lokaliziranih v kolagenskem genu. Dediščina je avtosomno prevladujoča.
skupine B. fenotipsko podobnih skupine A. Vendar kršitve v dihala niso tako izrazite, otroci z osteogenesis imperfecta živel več let. Obstajajo skrajšane cevaste kosti in spremembe strukture reber, vendar so njihovi zlomi redki pojav. Verjetno je dednost avtosomna recesivna.
skupina B. Taki primeri nepopolne osteogeneze so redki. Značilen zaradi mrtvorojenosti ali nastopa smrti v prvem mesecu. Pri bolnikih je opaziti majhno rast, finost cevastih kosti( zlasti diafizo), kostne lobanje brez osificiranja. Verjetno avtosomna recesivna vrsta dedovanja.
Bolniki s cepivom III nespodobne osteogeneze so redki. Med porodom se včasih pojavi zlom, telo novorojenčka se skrajša, medtem ko je masa v normalnih mejah. Karakteristična je deformacija prostih okončin v obliki O, ki je kifoskolioza. Vzrok smrti približno polovice bolnikov so različne spremembe v okostju in obtočnem sistemu. Izražena je osteoporoza, rast kosti v dolžini se prekine skupaj z njihovo oficifikacijo. V rastnih conah kostnega tkiva je opaziti neenakomerno kalcifikacijo, kar vodi v pojav popkov( tako imenovane "koruzne zrnje").Herednost ni točno znana.
IV vrsta nepopolne osteogeneze pri otrocih se najpogosteje kaže kot motnja v okostju. Ta vrsta ima veliko variabilnost osteopenije, število zlomov kosti, starost, modri sclero. Ponavadi s starostjo, se zmanjša število zlomov kosti, ki nastanejo žulji, po 30 letih pri nekaterih bolnikih ta vrsta imperfektna osteogeneza obravnave oslabljena. Glede na stanje dentina so bolniki razdeljeni v dve skupini: nekateri imajo ostre spremembe v dentinu( opalni zobmi), drugi pa v dentinu nimajo sprememb.
Nepravilno zdravljenje osteogeneze
Diagnoza nepopolne osteogeneze se najprej opira na klinično sliko bolezni. Za določitev natančne diagnoze je potrebno strokovno posvetovanje( endokrinolog, ortopedist ali genetik) in imenovanje laboratorijskih testov za izključitev drugih bolezni.
Laboratorijske metode vključujejo molekularno analizo kolagena in njegovo biokemijsko analizo, preučevanje materiala kožne biopsije. Instrumentalne metode študije vključujejo: rentgenski pregled( opredelitev osteopenija, različne zlome, ukrivljenost kosti, zlom hrbtenice, Intercalary kosti v lobanji), biopsijo kostnega denzitometrijo.
Glavni cilj zdravljenja osteogenesis imperfecta - zmanjšanje števila zlomov pri bolnikih, povečanje mineralizacijo kosti, da bi zmanjšali njihovo krhkost, povečanje kostne mase. Pomembna točka zdravljenja je prilagoditev otroka, pa tudi staršev do spremenjenega načina življenja zaradi bolezni. Tako se bodo izognili prihodnjim zlomom kosti.
Celoten potek zdravljenja je sestavljen iz:
- zdravila. Za boj proti osteogeneze imperfekta uporabljajo zdravila: difosfonate, rastnega hormona, vitamin D3, kalcijev glukonat, magnezijeve soli in kalijeve soli, ergokalciferola, kelatorjev, glicerofosfat.
- operativni poseg. Treba je odpraviti zlome, krepiti kosti, popraviti spremembe zaradi deformacije, protetike. Včasih kirurško zdravljenje ni ena operacija: zaradi rasti kosti predhodno vstavljeni zatiči zahtevajo zamenjavo.
Pri zdravljenju zlomov nanesemo prožne titanove palice, ki imajo hipoalergične lastnosti. Takšne palice so dobro pritrjene z zlomom, zato izgine potreba po dodatni imobilizaciji telesa. Običajna uporaba mavčnih oblog povzroča težave pri oskrbi, povzroča nove zlome. Prav tako je mogoče uporabiti različne cepiče, kostne plastike in druge kovinske strukture.
Glede na povečano krhkost kosti pri bolniku, v nekaterih primerih, ortopedski proizvajajo osteoclasis popraviti deformacije sprememb kosti, nato pa je ud fiksnih z mavčno povojem. Primeren tudi za imobilizacijo raztezanja telesa. Uporabljajo se tudi
poleg osnovne metode zdravljenja osteogenesis imperfecta: izvaja terapijo, fizioterapijo in osteosintezo. Opravili tečaj rehabilitacijo in psihološko podporo, kot člani pacienta in njegovih svojcev.
polno potek zdravljenja ni v celoti obvladovati tak hudo boleznijo, kot je osteogenesis imperfecta, da vse zgoraj navedene metode omogočajo le delno odpraviti simptome in lažji način življenja za bolnika, ne da bi to vplivalo na osnovni vzrok bolezni.