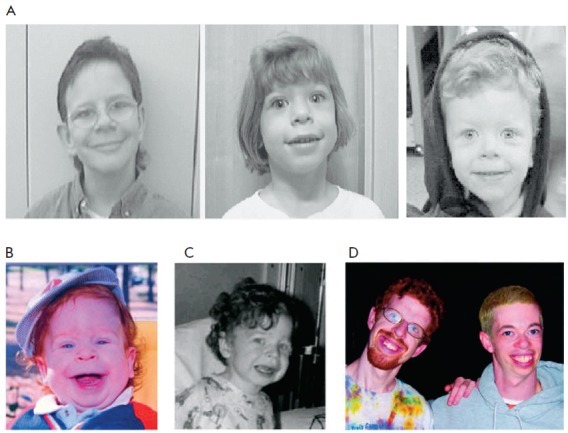
Lipoidose (Lysosomal lipid storage disease): hvad er det, eksempler
Opmærksomhed! Oplysningerne er kun til informationsformål og er ikke beregnet til at diagnosticere og ordinere behandling. Kontakt altid din speciallæge!
Indhold
- Hvad er lysosomale lagringssygdomme?
- Hvad er lipider?
- Hvordan nedarves lipoidose?
- Typer og symptomer på lysosomale lagringssygdomme
Hvad er lysosomale lagringssygdomme?
Lysosomale lagringssygdomme (lipoidose), er en gruppe af arvelige stofskiftesygdomme, hvor skadelige mængder af fedtstoffer (lipider) ophobes i forskellige celler og væv i kroppen.
Mennesker med disse lidelser laver enten ikke nok af et af de enzymer, der er nødvendige for at nedbryde (metabolisere) lipider, eller også producerer de enzymer, der ikke fungerer korrekt.
Over tid kan denne overdrevne ophobning af fedt føre til permanent skade på celler og væv, især i hjernen, det perifere nervesystem (nerver fra rygmarven til resten af legeme), lever, milt og knoglemarv.
Hvad er lipider?
Lipider er fedtlignende stoffer, som er vigtige dele af membranerne inde i og mellem celler og i myelinskeden, som dækker og beskytter nerverne. Lipider omfatter olier, fedtsyrer, voks, steroider (såsom kolesterol og østrogen) og andre relaterede forbindelser.
Disse fedtstoffer lagres naturligt i kroppens celler, organer og væv. Små kroppe i celler, kaldet lysosomer, omdanner eller metaboliserer regelmæssigt lipider og proteiner til mindre komponenter for at give energi til kroppen. Lidelser, hvor intracellulært materiale, som ikke kan metaboliseres, forbliver i lysosomer, kaldes lysosomale lagringssygdomme.
Ud over sygdomme forbundet med akkumulering af lipider omfatter andre lysosomale sygdomme forbundet med akkumulering mucolipidoser, hvor celler og væv ophobes. en overdreven mængde lipider med sukkermolekyler knyttet til dem, såvel som mucopolysaccharidoser, hvor en overdreven mængde stort, komplekst sukker ophobes molekyler.
Hvordan nedarves lipoidose?
Lysosomale lagringssygdomme nedarves fra en eller begge forældre, som bærer et defekt gen, der regulerer et specifikt lipid-metaboliserende enzym i en celleklasse i kroppen. De kan arves på to måder:
- Autosomal recessiv arv opstår, når begge forældre bærer og videregiver en kopi af det defekte gen, men ingen af forældrene er påvirket af lidelsen. Hvert barn født af disse forældre har en 25 procents chance for at arve begge kopier af det defekte gen, 50 procent sandsynligheden for, at han vil være en bærer, der ligner sine forældre, og en 25 procents chance for ikke at arve nogen kopi af den defekte gen. Børn af begge køn kan nedarves på en autosomal recessiv måde.
- X-bundet (eller kønsrelateret) recessiv arv opstår, når moderen bærer det berørte gen på X-kromosomet. X- og Y-kromosomerne er involveret i kønsbestemmelse. Kvinder har to X-kromosomer, mens mænd har et X-kromosom og et Y-kromosom. Sønner af kvindelige bærere har 50 procents chance for at arve og påvirke lidelsen, da sønner får et X-kromosom fra deres mor og et Y-kromosom fra deres far. Døtre har 50 procents chance for at arve det berørte X-kromosom fra deres mor og er bærere eller lettere påvirket. Berørte mænd overfører ikke sygdommen til deres sønner, men deres døtre vil være bærere og bærere af sygdommen.
Typer og symptomer på lysosomale lagringssygdomme
Gauchers sygdom - den mest almindelige af lysosomale sygdomme. Sygdommen skyldes en mangel på enzymet glucocerebrosidase, hvilket fører til ophobning af glucocerebrosid i mange væv. Fedtmateriale kan ophobes i hjernen, milten, leveren, nyrerne, lungerne og knoglemarven.
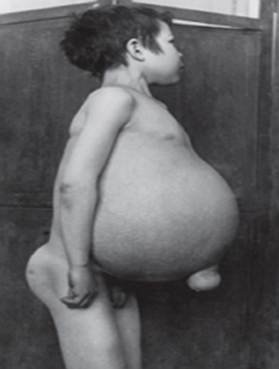
Symptomer kan omfatte hjerneskade, forstørrelse af milten og lever, nedsat leverfunktion, skeletlidelser og knogleskader, der kan forårsage smerter og brud, hævelse af lymfeknuder og (nogle gange) tilstødende led, oppustethed, brunlig hudfarve, anæmi, lavt antal blodplader og gule pletter på øjnene.
De personer, der er hårdest ramt, kan også være mere modtagelige for infektioner. Sygdommen rammer mænd og kvinder lige meget.
Gauchers sygdom har tre generelle kliniske undertyper:
- Type 1 (Eller nej neuronopatisk type) er den mest almindelige form for sygdommen i USA og Europa. Hjernen påvirkes ikke, men der kan være skader på lungerne og i sjældne tilfælde nyrerne. Symptomer kan begynde tidligt i livet eller i voksenalderen og omfatte en forstørret lever og alvorlig forstørrelse af milten, som kan føre til bristning og yderligere komplikationer. Skeletsvaghed og knoglesygdom kan være omfattende. Mennesker i denne gruppe får normalt let blå mærker på grund af det lave antal blodplader i deres blod. De kan også opleve træthed på grund af anæmi. Afhængigt af sygdommens opståen og sværhedsgrad kan personer med type 1 kan leve til voksenlivet. Mange syge har mild sygdom eller viser muligvis ingen symptomer. Selvom 1type Gauchers sygdom er almindelig blandt mennesker af Ashkenazi-jødisk arv og kan påvirke mennesker af alle etniske baggrunde.
- Type 2 (eller akut neuropatisk barndom Gauchers sygdom) begynder normalt inden for 3 måneder efter fødslen. Symptomerne omfatter omfattende og progressiv hjerneskade, spasticitet, krampeanfald, stive lemmer, forstørret lever (lever hepatomegali) og milt (splenomegali), unormale øjenbevægelser og dårlig evne til at sutte og synke. Berørte børn dør normalt før 2 års alderen.
- Type 3 (kronisk neuronopatisk form) kan begynde når som helst i barndommen eller endda i voksenalderen. Det er karakteriseret ved langsomt fremadskridende, men mindre alvorlige neurologiske symptomer sammenlignet med akut Gauchers sygdom 2 typer. Vigtigste symptomer omfatter øjenbevægelsesforstyrrelser, kognitive underskud, dårlig koordination, anfald, forstørrelse af milten og/eller leveren, skeletlidelser, blodsygdomme inklusive anæmi og problemer med vejrtrækning. Næsten alle med Gauchers sygdom 3 typer, som modtager enzymerstatningsterapi vil blive voksen.
Til patienter 1. og 3. type Enzymerstatningsterapi, givet intravenøst hver anden uge, kan dramatisk reducere størrelsen af leveren og milten, reducere skeletabnormiteter og vende andre manifestationer. En vellykket knoglemarvstransplantation behandler de neurologiske manifestationer af sygdommen. Denne procedure indebærer imidlertid betydelige risici og udføres sjældent hos personer med Gauchers sygdom.
I sjældne tilfælde kan det være nødvendigt at operere for at fjerne hele eller dele af milten (hvis en person har et meget lavt antal blodplader, eller når et forstørret organ i høj grad påvirker personens komfort). Blodtransfusion kan gavne nogle mennesker med anæmi. Andre kan have behov for en ledudskiftningsoperation for at forbedre mobiliteten og livskvaliteten. Der findes i øjeblikket ingen effektiv behandling af de hjerneskader, der kan opstå hos mennesker med Gauchers sygdom type 2 og 3.
Niemann-Pick sygdom er en gruppe autosomale recessive lidelser forårsaget af ophobning af fedt og kolesterol i cellerne i leveren, milten, knoglemarven, lungerne og i nogle tilfælde hjernen.
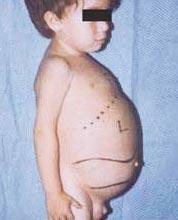
Neurologiske komplikationer kan omfatte ataksi (manglende muskelkoordination, der kan påvirke gang, skrivning og spisning, blandt andre funktioner), øjenlammelse, hjernedegeneration, indlæringsproblemer, spasticitet, besvær med at spise og synke, sløret tale, tab af muskeltonus, overfølsomhed over for berøring og en vis uklarhed i hornhinden på grund af overdreven ophobning materialer. Hos 50 % af de ramte mennesker udvikles en karakteristisk kirsebærrød glorie omkring nethindens centrum, som kan ses af en læge med et særligt instrument.
Niemann-Picks sygdom er klassificeret i tre generelle undertyper:
- Type A, den mest alvorlige form, begynder i den tidlige barndom. Babyer ser normale ud ved fødslen, men efter 6 måneder udvikles der dyb hjerneskade, en forstørret lever og milt og hævede lymfeknuder og noder under huden (xanthomas). Milten kan blive 10 gange dens normale størrelse og kan briste og forårsage blødning. Disse børn bliver gradvist svagere, mister motorisk funktion, kan blive anæmiske og er tilbøjelige til tilbagevendende infektioner. De lever sjældent mere end 18 måneder. Denne form for sygdommen er mest almindelig i jødiske familier.
- Type B(eller juvenil debut) påvirker normalt ikke hjernen, men de fleste børn udvikler sig ataksi, beskadigelse af nerverne, der forlader rygmarven (perifer neuropati), og lungebesvær, der udvikler sig med alderen. Forstørrelse af lever og milt er typisk for unge. Folk med type B kan leve relativt længe, men mange kræver livslang iltbehandling på grund af lungeskader. Niemann-Pick type A og B er resultatet af en ophobning af et fedtstof kaldet sphingomyelin på grund af en mangel på et enzym kaldet sphingomyelinase.
- Type C kan dukke op tidligt i livet eller udvikle sig i ungdomsårene eller endda voksenalderen. Niemann-Pick sygdomtype C skyldes ikke sphlingomyelinase-mangel, men af mangel på NPC1- eller NPC2-proteiner. Som et resultat ophobes forskellige lipider og især kolesterol i nerveceller og forårsager deres funktionsfejl. Hjerneinvolvering kan være omfattende, hvilket resulterer i manglende evne til at se op og ned, besvær med at gå og synke, progressivt høretab og progressivt demens. Folk med type C har kun mild forstørrelse af milt og lever. Forventet levetid for mennesker med type C varierer betydeligt. Nogle mennesker dør i barndommen, mens andre, der er mindre ramt, også kan leve i voksenalderen.
Der er i øjeblikket ingen kur mod Niemann-Picks sygdom. Behandlingen er støttende. Børn dør normalt af infektion eller progressivt neurologisk tab. Der har været tilfælde af knoglemarvstransplantation hos flere patienter med type B med blandede resultater.
Fabrys sygdomogså kendt som alfa-galactosidase-A-mangelforårsager ophobning af fedtstoffer i det autonome nervesystem (den del af nervesystemet, der kontrollerer ufrivillige funktioner såsom vejrtrækning og hjerteslag), i øjne, nyrer og kardiovaskulære system.
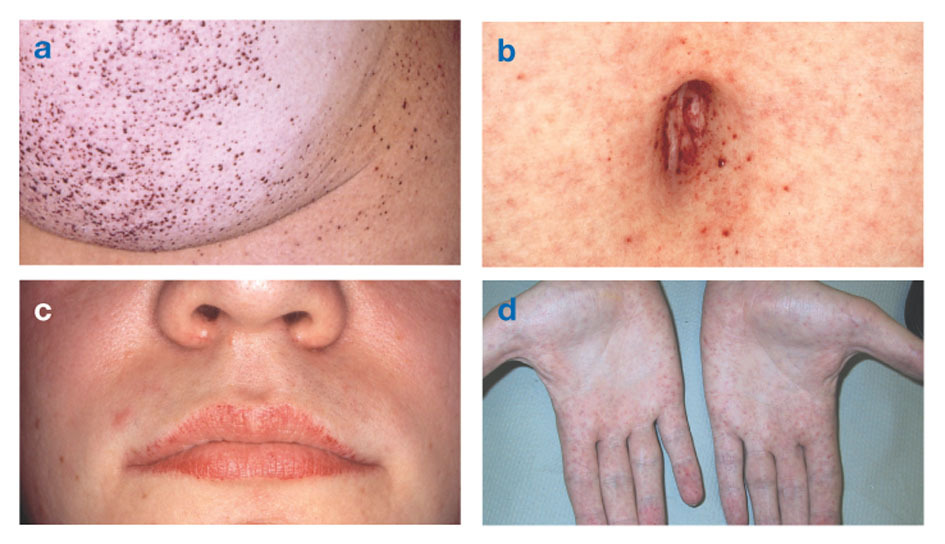
Fabrys sygdom er den eneste X-bundne lipidsygdom. Sygdommen rammer hovedsageligt mænd, selvom en mildere og mere varierende form forekommer hos kvinder. Sjældent har berørte kvinder alvorlige symptomer, der ligner dem, der ses hos mænd med lidelsen. Begyndelsen af symptomer opstår normalt i barndommen eller ungdommen.
Neurologiske tegn omfatter brændende smerter i arme og ben, der bliver værre i varmt vejr eller efter træning, samt ophobning af overskydende materiale i de gennemsigtige lag af hornhinden (hvilket fører til uklarhed, men uden ændringer i synet). Fedtophobning i væggene i blodkarrene kan forstyrre cirkulationen og sætte en person i fare slag eller hjerteanfald. Andre symptomer omfatter et forstørret hjerte, progressivt Nyresvigtfører til nyresygdom i slutstadiet, mave-tarmproblemer, nedsat svedtendens og feber.
Angiokeratomer (små, ikke-kræftfremkaldende, rødlilla hævede pletter på huden) kan udvikle sig i underkroppen og blive mere almindelige med alderen.
Mennesker med Fabrys sygdom dør ofte for tidligt af komplikationer fra hjertefejl, nyresvigt eller slagtilfælde. Medicin som phenytoin og carbamazepin ordineres ofte til at behandle smerten, der ledsager Fabrys sygdom, men ikke for at helbrede den. Metoclopramid eller Lipisorb (kosttilskud) kan lindre gastrointestinale forstyrrelser forekommer ofte hos personer med Fabrys sygdom, og nogle mennesker kan have behov for en nyretransplantation eller dialyse. Enzymerstatning kan reducere opbevaring, lindre smerter og bevare organfunktionen hos nogle mennesker med Fabrys sygdom.
Farbers sygdom også kendt som Farbers lipogranulomatose, beskriver en gruppe sjældne autosomale recessive lidelser, der forårsager ophobning af fedtstoffer i led, væv og centralnervesystemet. Det rammer både mænd og kvinder. Sygdommen begynder normalt i den tidlige barndom, men kan opstå senere i livet.
Børn med klassisk Farbers sygdom udvikler sig neurologiske symptomer, som kan omfatte øget søvnighed og sløvhed og problemer med synke. Lever, hjerte og nyrer kan også blive påvirket. Andre symptomer kan omfatte ledkontrakturer (kronisk forkortelse af muskler eller sener omkring leddene), opkastning, gigt, forstørrelse lymfeknuder, ledbetændelse, hæshed og klumper under huden, der bliver tykkere omkring leddene, efterhånden som den skrider frem sygdomme.
Berørte personer med åndedrætsbesvær kan have behov for at få indsat en åndedrætsslange. De fleste børn med denne tilstand dør ved 2 års alderen, normalt fra lungesygdomme. I en af de mest alvorlige former for sygdommen kan en forstørret lever og milt diagnosticeres kort efter fødslen. Babyer født med denne form for sygdommen dør normalt inden for 6 måneder.
Farbers sygdom er forårsaget af en mangel på et enzym kaldet ceramidase. Der er i øjeblikket ingen specifik behandling for Farbers sygdom. Kortikosteroider kan ordineres for at lindre smerter. Knoglemarvstransplantation kan reducere granulomer (små masser af betændt væv) hos mennesker med mild inflammation eller hos patienter uden komplikationer i lunge- eller nervesystemet. Hos ældre mennesker kan granulomer krympes eller kirurgisk fjernes.
Gangliosidose består af to forskellige grupper af genetiske sygdomme. Begge er autosomalt recessive og påvirker mænd og kvinder lige meget.
GM1 gangliosidose.
GM1 gangliosidose er forårsaget af en mangel på enzymet beta-galactosidase, hvilket resulterer i en unormal ophobning af sure lipidmaterialer, især i nerveceller i det centrale og perifere nervesystem systemer. GM1 gangliosidose har tre kliniske manifestationer:
- GM1 (den mest alvorlige undertype, der opstår kort efter fødslen) kan omfatte neurodegeneration, kramper, forstørret lever og milt, grove ansigtstræk, skelet-uregelmæssigheder, ledstivhed, oppustethed, muskelsvaghed, overdreven forskrækkelsesrespons og problemer med gang. Omkring halvdelen af de ramte udvikler kirsebærrøde pletter i øjet. Børn kan være døve og blinde ved 1 års alderen og dør ofte ved 3 års alderen af hjertekomplikationer eller lungebetændelse.
- Sen barn GM1 gangliosidose begynder normalt i alderen 1 og 3 år. Neurologiske symptomer omfatter ataksi, anfald, demens og talebesvær.
- GM1 gangliosidose udvikler sig mellem 3 og 30 år. Symptomerne omfatter nedsat muskelmasse (muskelatrofi), neurologiske komplikationer, der er mindre alvorlige og udvikler sig langsommere end andre former. lidelser, hornhindeopacitet hos nogle mennesker og vedvarende muskelsammentrækninger, der forårsager vridninger og gentagne bevægelser eller unormale stillinger (dystoni). Angiokeratomer kan udvikle sig i den nederste del af kroppen. Størrelsen af leveren og milten er normal hos de fleste berørte mennesker.
GM2 gangliosidose.
GM2 gangliosidose får også kroppen til at akkumulere overskydende sure fedtstoffer i væv og celler, primært nerveceller. Disse lidelser skyldes en mangel på enzymet beta-hexosaminidase. GM2 lidelser omfatter:
- Tay-Sachs sygdom (også kendt som GM2 gangliosidosis variant B) og dens variantformer er forårsaget af en mangel på enzymet hexosaminidase A. Berørte børn udvikler sig normalt i løbet af de første par måneder af livet. Symptomer begynder ved 6-måneders alderen og omfatter progressivt tab af mental kapacitet, demens, nedsat øjenkontakt, øget en forskrækkelsesreaktion på støj, progressivt høretab, der fører til døvhed, synkebesvær, blindhed, kirsebærrøde pletter på nethinden og nogle lammelse. Anfald kan begynde i det andet år af et barns liv. Babyer kan i sidste ende blive afhængige af sonden og dør ofte ved 4-års alderen af tilbagevendende infektioner. Desværre er der ingen særlig behandling. Antikonvulsiva kan i begyndelsen kontrollere anfald. Andre støttende behandlinger omfatter korrekt ernæring og hydrering og metoder til at holde luftvejene åbne. En sjælden form for lidelsen kaldet sent opstået Tay-Sachs sygdom, forekommer hos mennesker mellem 20 og 30 år og er karakteriseret ved en ustabil gangart og progressiv neurologisk forringelse.
- Sandhoffs sygdom (Option AB) er en alvorlig form for Tay-Sachs sygdom. Debut forekommer normalt ved 6 måneders alderen og er ikke begrænset til nogen etnisk gruppe. Neurologiske tegn kan omfatte progressiv forringelse af centralnervesystemet, motorik svaghed, tidlig blindhed, alvorlig bange reaktion på lyd, spasticitet, chok eller trækninger muskler (myoklonus), epilepsi, et unormalt forstørret hoved (makrocefali) og røde pletter i øjet. Andre symptomer kan omfatte hyppige luftvejsinfektioner, hjertemislyd, dukkelignende ansigtstræk og forstørrelse af lever og milt. Der er ingen specifik behandling for Sandhoffs sygdom. Som med Tay-Sachs sygdom omfatter støttende pleje at holde luftvejene åbne hele tiden samt korrekt ernæring og hydrering. Antikonvulsiva kan i begyndelsen kontrollere epilepsi (anfald).
Krabbes sygdom(også kendt som kuglecelleleukodystrofi og galactosylceramid lipidose) er en autosomal recessiv sygdom forårsaget af en mangel på enzymet galactocerebrosidase. Sygdommen rammer oftest spædbørn, der debuterer før 6 måneders alderen, men kan forekomme i ungdomsårene eller i voksenalderen.
Ophobningen af ufordøjet fedt påvirker væksten af nervens beskyttende kappe (myelinskeden) og forårsager alvorlig svækkelse af mentale og motoriske færdigheder. Andre symptomer omfatter muskelsvaghed, nedsat evne til at strække musklerne (hypotoni), muskelspændinger (spasticitet), pludseligt chok eller trækninger i lemmerne (myokloniske anfald), irritabilitet, uforklarlig feber, døvhed, blindhed, lammelse og synkebesvær. Langsigtet vægttab kan også forekomme.
Sygdommen kan diagnosticeres ved enzymtestning og identifikation af dens karakteristiske cellegruppering ind i globoide kroppe i hjernens hvide substans, demyelinisering af nerver og degeneration og ødelæggelse af hjerneceller. Hos spædbørn er sygdommen normalt dødelig før 2 års alderen. Mennesker med en mere fremskreden form for sygdom har et mildere sygdomsforløb og lever væsentligt længere.
Der er ikke udviklet nogen specifik behandling for Krabbes sygdom, selvom tidlig knoglemarvstransplantation kan hjælpe nogle mennesker. Mennesker med en mere fremskreden form for sygdom har et mildere sygdomsforløb og lever væsentligt længere.
Metakromatisk leukodystrofi, eller MLD, er en gruppe lidelser, der er præget af ophobning af fedtstoffer i det hvide stof i centralnervesystemet og i perifere nerver og til en vis grad i nyrerne. I lighed med Krabbes sygdom påvirker MLD myelinet, som dækker og beskytter nerverne. Denne autosomale recessive lidelse er forårsaget af en mangel på enzymet arylsulfatase A. Denne lidelse påvirker både mænd og kvinder.
Metakromatisk leukodystrofi har tre karakteristiske former: sent spædbarn, juvenil og voksen.
- Late Infant MLD starter normalt mellem 12 og 20 måneder efter fødslen. Børn kan virke normale i starten, men har svært ved at gå og har en tendens til at falde, ledsaget af periodiske smerter i arme og ben. progressivt tab af synet, der fører til blindhed, udviklingsforsinkelser og tab af tidligere erhvervede milepæle, svækket synke, anfald og demens op til 2 flere år. Børn udvikler også progressiv muskelsvind og svaghed og mister til sidst deres evne til at gå. De fleste børn med denne form for lidelse dør ved 5 års alderen.
- Juvenile MLD begynder normalt mellem 3 og 10 år. Symptomerne omfatter nedsat skolepræstation, mental svækkelse, ataksi, anfald og demens. Symptomerne udvikler sig, og døden indtræffer 10-20 år efter sygdommens opståen.
- Voksen form. Symptomer voksne debut efter 16 år og kan omfatte ataksi, kramper, unormale rystelser i lemmerne (rysten), nedsat koncentrationsevne, depression, psykiske lidelser og demens. Døden indtræffer normalt 6-14 år efter symptomernes begyndelse.
MLD kan ikke helbredes. Behandlingen er symptomatisk og understøttende. Knoglemarvstransplantation kan i nogle tilfælde forsinke udviklingen af sygdommen. Der er gjort betydelige fremskridt med genterapi i dyremodeller med MLD og i kliniske forsøg.
Wolmans sygdomogså kendt som lysosomal syrelipase-mangeler en alvorlig lipidlagringsforstyrrelse, der normalt er dødelig ved 1 års alderen. Denne autosomale recessive lidelse er karakteriseret ved en ophobning af kolesterolestere (normalt transportformen af kolesterol) og triglycerider (den kemiske form, hvori fedtstoffer findes i kroppen), som kan ophobes betydeligt og forårsage celleskade og stoffer.
Både mænd og kvinder lider af denne lidelse. Spædbørn er normale og aktive ved fødslen, men udvikler hurtigt progressiv mental svækkelse, en forstørret lever og stærkt forstørret milt, oppustethed, mave-tarmproblemer. gulsot, anæmi, opkastning og kalkaflejringer i binyrernefår dem til at hærde.
En anden type lysosomal syrelipase-mangel er sygdom til opbevaring af kolesterolester. Denne ekstremt sjældne sygdom opstår som følge af lagring af kolesterolestere og triglycerider i blodceller, lymfe og lymfoidt væv. Hos børn i voksenalderen forstørres leveren, hvilket fører til skrumpelever og kronisk leversvigt. Børn kan også have kalkaflejringer i binyrerne, og gulsot kan udvikle sig i de senere stadier af sygdommen.
I øjeblikket undersøges spørgsmålet om enzymerstatning aktivt både i Wolmans sygdom og i akkumulering af en kolesterolester.
Opmærksomhed! Oplysningerne er kun til informationsformål og er ikke beregnet til at diagnosticere og ordinere behandling. Kontakt altid din speciallæge!