Lipoidose (Lysosomale Lipidspeicherkrankheit): Was ist das, Beispiele
Beachtung! Die Informationen dienen nur zu Informationszwecken und dienen nicht der Diagnose und Verschreibung einer Behandlung. Konsultieren Sie immer Ihren Facharzt!
Inhalt
- Was sind lysosomale Speicherkrankheiten?
- Was sind Lipide?
- Wie werden Lipoidose vererbt?
- Arten und Symptome von lysosomalen Speicherkrankheiten
Was sind lysosomale Speicherkrankheiten?
Lysosomale Speicherkrankheiten (Lipidose), sind eine Gruppe erblicher Stoffwechselerkrankungen, bei denen sich schädliche Mengen an Fettstoffen (Lipiden) in verschiedenen Zellen und Geweben des Körpers anreichern.
Menschen mit diesen Störungen produzieren entweder nicht genug von einem der Enzyme, die zum Abbau (metabolisieren) von Lipiden benötigt werden, oder sie produzieren Enzyme, die nicht richtig funktionieren.
Im Laufe der Zeit kann diese übermäßige Ansammlung von Fett zu dauerhaften Schäden an Zellen und Gewebe führen, insbesondere im Gehirn, peripheres Nervensystem (Nerven vom Rückenmark bis zum Rest des Karosserie), Leber, Milz und Knochenmark.
Was sind Lipide?
Lipide sind fettähnliche Substanzen, die wichtige Bestandteile der Membranen innerhalb und zwischen den Zellen sowie in der Myelinscheide sind, die die Nerven bedeckt und schützt. Lipide umfassen Öle, Fettsäuren, Wachse, Steroide (wie Cholesterin und Östrogen) und andere verwandte Verbindungen.
Diese Fettstoffe werden auf natürliche Weise in den Zellen, Organen und Geweben des Körpers gespeichert. Winzige Körper in Zellen, die als Lysosomen bezeichnet werden, wandeln regelmäßig Lipide und Proteine in kleinere Komponenten um oder verstoffwechseln sie, um dem Körper Energie zu liefern. Störungen, bei denen intrazelluläres Material, das nicht metabolisiert werden kann, in Lysosomen verbleibt, werden als lysosomale Speicherkrankheiten bezeichnet.
Neben Krankheiten, die mit der Ansammlung von Lipiden verbunden sind, gehören zu anderen lysosomalen Erkrankungen, die mit einer Ansammlung verbunden sind, Mukolipidosen, bei denen sich Zellen und Gewebe ansammeln eine übermäßige Menge an Lipiden mit daran gebundenen Zuckermolekülen sowie Mucopolysaccharidosen, in denen sich eine übermäßige Menge an großen, komplexen Zuckern ansammelt Moleküle.
Wie werden vererbt? Lipoidose?
Lysosomale Speicherkrankheiten werden von einem oder beiden Elternteilen vererbt, die ein defektes Gen tragen, das ein bestimmtes lipidmetabolisierendes Enzym in einer Klasse von Körperzellen reguliert. Sie können auf zwei Arten vererbt werden:
- Autosomal-rezessive Vererbung tritt auf, wenn beide Elternteile eine Kopie des defekten Gens tragen und weitergeben, aber keiner der Elternteile von der Störung betroffen ist. Jedes Kind dieser Eltern hat eine 25-prozentige Chance, beide Kopien des defekten Gens zu erben, 50 Prozent die Wahrscheinlichkeit, dass er ein ähnlicher Träger wie seine Eltern ist, und eine 25-prozentige Chance, keine Kopie des defekten zu erben Gen. Kinder beiderlei Geschlechts können autosomal-rezessiv vererbt werden.
- X-chromosomale (oder geschlechtsbezogene) rezessive Vererbung tritt auf, wenn die Mutter das betroffene Gen auf dem X-Chromosom trägt. Die X- und Y-Chromosomen sind an der Geschlechtsbestimmung beteiligt. Frauen haben zwei X-Chromosomen, Männer ein X-Chromosom und ein Y-Chromosom. Söhne weiblicher Träger haben eine 50-prozentige Chance, die Krankheit zu erben und zu beeinflussen, da Söhne ein X-Chromosom von ihrer Mutter und ein Y-Chromosom von ihrem Vater erhalten. Töchter haben eine 50-prozentige Chance, das betroffene X-Chromosom von ihrer Mutter zu erben und sind Trägerinnen oder leicht betroffen. Betroffene Männer übertragen die Krankheit nicht auf ihre Söhne, aber ihre Töchter werden Träger und Träger der Krankheit sein.
Arten und Symptome von lysosomalen Speicherkrankheiten
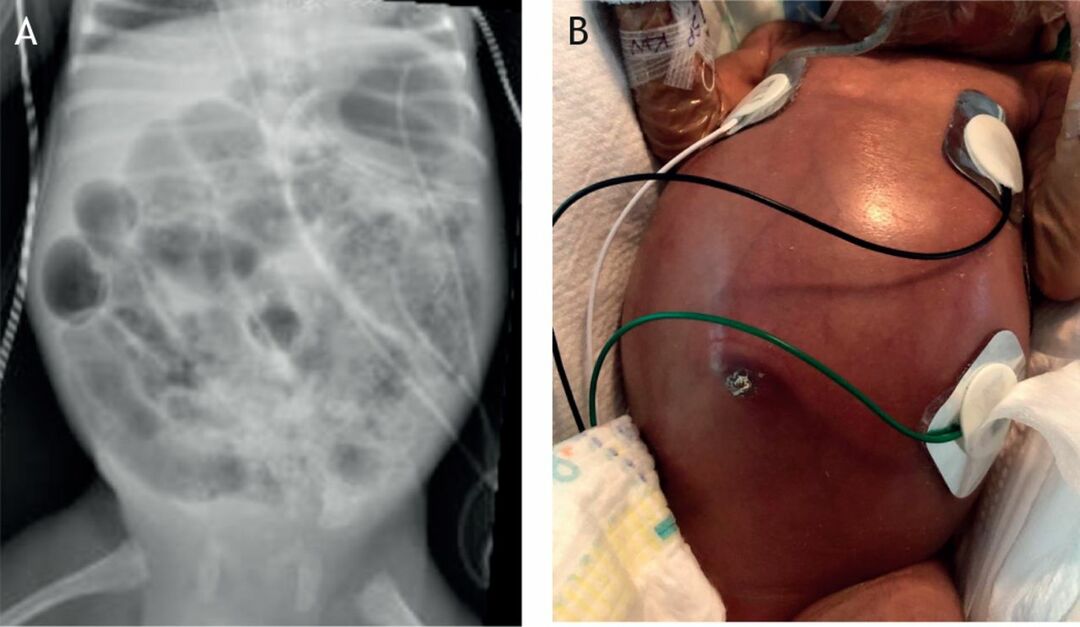
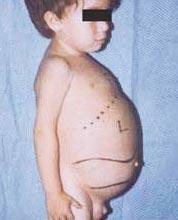
Gaucher-Krankheit - die häufigste der lysosomalen Erkrankungen. Die Krankheit wird durch einen Mangel des Enzyms Glucocerebrosidase verursacht, der in vielen Geweben zur Anreicherung von Glucocerebrosid führt. Fettes Material kann sich im Gehirn, in der Milz, in der Leber, in den Nieren, in der Lunge und im Knochenmark ansammeln.
Symptome können Hirnschäden, Vergrößerung der Milz und Leber, eingeschränkte Leberfunktion, Skeletterkrankungen und Knochenschäden, die Schmerzen und Frakturen verursachen können, Schwellungen der Lymphknoten und (manchmal) angrenzender Gelenke, Blähungen, bräunlicher Hautton, Anämie, niedrige Thrombozytenzahl und gelbe Flecken auf den Augen.
Die am schwersten betroffenen Personen können auch anfälliger für Infektionen sein. Die Krankheit betrifft Männer und Frauen gleichermaßen.
Morbus Gaucher hat drei allgemeine klinische Subtypen:
- Typ 1 (Oder Nein neuronopathisch Typ) ist die häufigste Form der Krankheit in den Vereinigten Staaten und Europa. Das Gehirn ist nicht betroffen, es kann jedoch zu Schäden an der Lunge und in seltenen Fällen auch an den Nieren kommen. Die Symptome können früh im Leben oder im Erwachsenenalter beginnen und umfassen eine vergrößerte Leber und eine starke Vergrößerung der Milz, die zu einer Ruptur und zusätzlichen Komplikationen führen können. Skelettschwäche und Knochenerkrankungen können ausgedehnt sein. Menschen in dieser Gruppe bekommen aufgrund der niedrigen Thrombozytenzahl in ihrem Blut normalerweise leicht blaue Flecken. Sie können auch aufgrund von Anämie ermüden. Je nach Ausbruch und Schwere der Erkrankung können Menschen mit Typ 1 erwachsen werden kann. Viele Betroffene haben eine leichte Erkrankung oder zeigen keine Symptome. Obwohl 1Typ Die Gaucher-Krankheit ist bei Menschen mit aschkenasischer jüdischer Herkunft weit verbreitet und kann Menschen aller ethnischen Hintergründe betreffen.
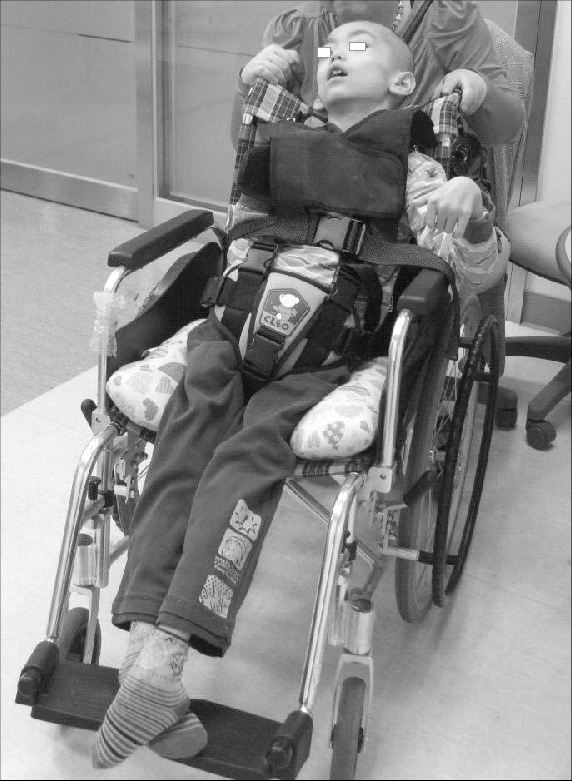
- Typ 2 (oder akute Neuropathie im Kindesalter Gaucher-Krankheit) beginnt in der Regel innerhalb von 3 Monaten nach der Geburt. Zu den Symptomen zählen ausgedehnte und fortschreitende Hirnschäden, Spastik, Krampfanfälle, steife Gliedmaßen, vergrößerte Leber (Leberhepatomegalie) und Milz (Splenomegalie), abnorme Augenbewegungen und schlechte Saug- und Schluckfähigkeit. Betroffene Kinder sterben in der Regel vor dem 2. Lebensjahr.
- Typ 3 (chronisch neuronopathisch Form) kann jederzeit in der Kindheit oder sogar im Erwachsenenalter beginnen. Sie zeichnet sich durch langsam fortschreitende, aber weniger schwere neurologische Symptome im Vergleich zur akuten Gaucher-Krankheit aus 2 Typen. Hauptsymptome sind Augenbewegungsstörungen, kognitive Defizite, schlechte Koordination, Krampfanfälle, Vergrößerung der Milz und/oder Leber, Skeletterkrankungen, Bluterkrankungen einschließlich Anämie und Probleme mit Atmung. Fast alle mit Gaucher-Krankheit 3 Arten, wer eine Enzymersatztherapie erhält, wird das Erwachsenenalter erreichen.
Für Patienten 1. und 3. Typ Eine Enzymersatztherapie, die alle zwei Wochen intravenös verabreicht wird, kann die Größe von Leber und Milz dramatisch reduzieren, Skelettanomalien reduzieren und andere Manifestationen rückgängig machen. Eine erfolgreiche Knochenmarktransplantation behandelt die neurologischen Manifestationen der Krankheit. Dieses Verfahren birgt jedoch erhebliche Risiken und wird bei Personen mit Gaucher-Krankheit selten durchgeführt.
In seltenen Fällen kann eine Operation erforderlich sein, um die Milz ganz oder teilweise zu entfernen (wenn eine Person eine sehr niedrige Thrombozytenzahl hat oder wenn ein vergrößertes Organ das Wohlbefinden der Person stark beeinträchtigt). Bluttransfusionen können einigen Menschen mit Anämie zugute kommen. Andere benötigen möglicherweise eine Gelenkersatzoperation, um die Mobilität und Lebensqualität zu verbessern. Es gibt derzeit keine wirksame Behandlung für die Hirnschäden, die bei Menschen mit Morbus Gaucher Typ 2 und 3 auftreten können.
Niemann-Pick-Krankheit ist eine Gruppe von autosomal-rezessiven Erkrankungen, die durch die Ansammlung von Fett und Cholesterin in den Zellen der Leber, Milz, des Knochenmarks, der Lunge und in einigen Fällen des Gehirns verursacht werden.
Neurologische Komplikationen können Ataxie (Mangel an Muskelkoordination, die das Gehen, Schreiben und Essen beeinträchtigen kann, unter anderem), Augenlähmung, Gehirndegeneration, Lernschwierigkeiten, Spastik, Schwierigkeiten beim Füttern und Schlucken, undeutlich Sprache, Verlust des Muskeltonus, Überempfindlichkeit gegenüber Berührungen und eine gewisse Hornhauttrübung aufgrund übermäßiger Akkumulation Materialien. Bei 50% der Betroffenen entwickelt sich um das Zentrum der Netzhaut ein charakteristischer kirschroter Lichthof, der von einem Arzt mit einem speziellen Instrument gesehen werden kann.
Die Niemann-Pick-Krankheit wird in drei allgemeine Subtypen eingeteilt:
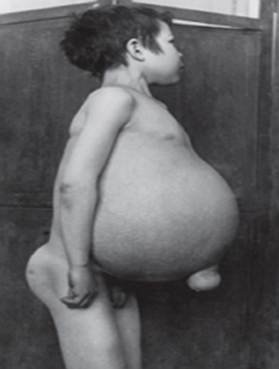
- Tippe A, die schwerste Form, beginnt in der frühen Kindheit. Babys erscheinen bei der Geburt normal, aber nach 6 Monaten entwickeln sich tiefe Hirnschäden, eine vergrößerte Leber und Milz sowie geschwollene Lymphknoten und Knoten unter der Haut (Xanthome). Die Milz kann das Zehnfache ihrer normalen Größe annehmen und reißen, was zu Blutungen führen kann. Diese Kinder werden zunehmend schwächer, verlieren motorische Funktionen, können anämisch werden und sind anfällig für wiederkehrende Infektionen. Sie leben selten länger als 18 Monate. Diese Form der Krankheit tritt am häufigsten in jüdischen Familien auf.
- Typ B(oder juveniler Beginn) hat normalerweise keinen Einfluss auf das Gehirn, aber die meisten Kinder entwickeln Ataxia, Schädigung der Nerven, die das Rückenmark verlassen (periphere Neuropathie) und Lungenbeschwerden, die mit dem Alter fortschreiten. Die Vergrößerung von Leber und Milz ist typisch für Jugendliche. Leute mit Typ B können relativ lange leben, aber viele benötigen aufgrund von Lungenschäden eine lebenslange Sauerstofftherapie. Die Niemann-Pick-Typen A und B sind das Ergebnis einer Ansammlung einer Fettsubstanz namens Sphingomyelin aufgrund eines Mangels an einem Enzym namens Sphingomyelinase.
- Typ C kann früh im Leben auftreten oder sich während der Adoleszenz oder sogar im Erwachsenenalter entwickeln. Niemann-Pick-KrankheitTyp C wird nicht durch einen Sphlingomyelinase-Mangel, sondern durch einen Mangel an NPC1- oder NPC2-Proteinen verursacht. Dadurch reichern sich verschiedene Lipide und insbesondere Cholesterin in Nervenzellen an und verursachen deren Fehlfunktion. Die Beteiligung des Gehirns kann umfangreich sein, was zu einer Unfähigkeit, nach oben und unten zu schauen, zu Schwierigkeiten beim Gehen und Schlucken, fortschreitendem Hörverlust und fortschreitendem. führt Demenz. Leute mit Typ C nur eine leichte Vergrößerung von Milz und Leber haben. Lebenserwartung von Menschen mit Typ C sehr unterschiedlich. Manche Menschen sterben im Kindesalter, während andere, die weniger betroffen sind, auch im Erwachsenenalter leben können.
Die Niemann-Pick-Krankheit ist derzeit nicht heilbar. Die Behandlung ist unterstützend. Kinder sterben in der Regel an einer Infektion oder einem fortschreitenden neurologischen Verlust. Es gab Fälle von Knochenmarktransplantationen bei mehreren Patienten mit Typ B mit gemischten Ergebnissen.
Morbus Fabryauch bekannt als Alpha-Galactosidase-A-Mangel, verursacht die Ansammlung von Fettstoffen im autonomen Nervensystem (dem Teil des Nervensystems, der steuert unwillkürliche Funktionen wie Atmung und Herzschlag), in Augen, Nieren und Herz-Kreislauf System.
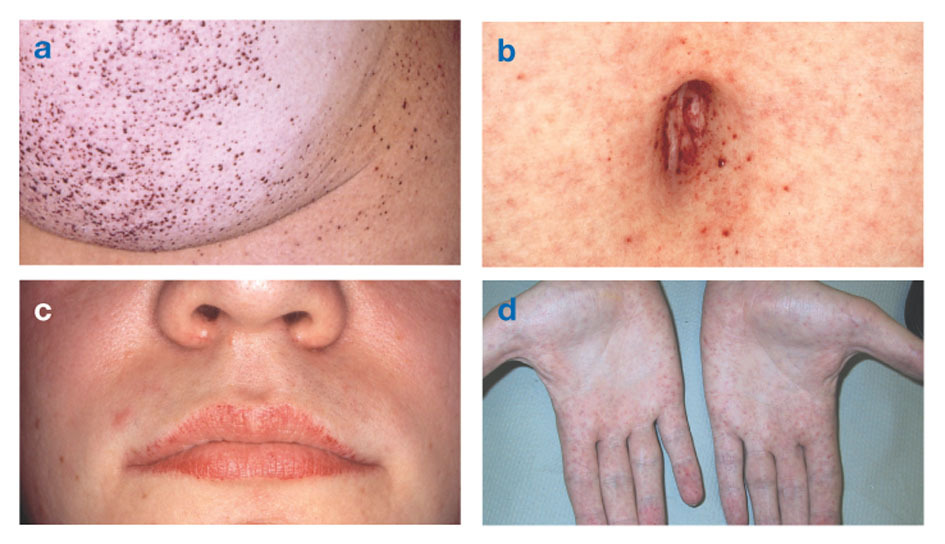
Morbus Fabry ist die einzige X-chromosomale Lipiderkrankung. Die Krankheit betrifft hauptsächlich Männer, obwohl bei Frauen eine mildere und variablere Form auftritt. In seltenen Fällen haben betroffene Frauen schwere Symptome, die denen ähnlich sind, die bei Männern mit der Erkrankung beobachtet werden. Die Symptome treten meist im Kindes- oder Jugendalter auf.
Neurologische Symptome sind brennende Schmerzen in Armen und Beinen, die sich bei heißem Wetter oder danach verschlimmern Übung, sowie die Ansammlung von überschüssigem Material in den transparenten Schichten der Hornhaut (was zu einer Trübung führt, aber ohne Sehveränderungen). Fettansammlungen in den Wänden der Blutgefäße können die Durchblutung stören und eine Person gefährden Schlaganfall oder Herzinfarkt. Andere Symptome sind ein vergrößertes Herz, progressive NierenversagenDies führt zu Nierenerkrankungen im Endstadium, Magen-Darm-Problemen, vermindertem Schwitzen und Fieber.
Angiokeratome (kleine, nicht krebsartige, rötlich-violette erhabene Flecken auf der Haut) können sich im unteren Rumpf entwickeln und mit zunehmendem Alter häufiger werden.
Menschen mit Morbus Fabry sterben oft vorzeitig an Komplikationen von Herzfehler, Nierenversagen oder Schlaganfall. Medikamente wie Phenytoin und Carbamazepin werden oft verschrieben, um die Schmerzen zu behandeln, die mit Morbus Fabry einhergehen, aber nicht, um sie zu heilen. Metoclopramid oder Lipisorb (Nahrungsergänzungsmittel) können Magen-Darm-Beschwerden lindern, die tritt häufig bei Patienten mit Morbus Fabry auf, und einige Patienten benötigen möglicherweise eine Nierentransplantation oder Dialyse. Enzymersatz kann bei einigen Menschen mit Morbus Fabry die Lagerung reduzieren, Schmerzen lindern und die Organfunktion erhalten.
Morbus Farber auch bekannt als Farber-Lipogranulomatose, beschreibt eine Gruppe seltener autosomal-rezessiv vererbter Erkrankungen, die zur Ansammlung von Fett in Gelenken, Geweben und im zentralen Nervensystem führen. Es betrifft sowohl Männer als auch Frauen. Die Krankheit beginnt in der Regel in der frühen Kindheit, kann aber auch später im Leben auftreten.
Kinder mit klassischem Morbus Farber entwickeln neurologische Symptome, die verstärkte Schläfrigkeit und Lethargie umfassen können, sowie Probleme mit schlucken. Auch Leber, Herz und Nieren können betroffen sein. Andere Symptome können Gelenkkontrakturen (chronische Verkürzung von Muskeln oder Sehnen um die Gelenke), Erbrechen, Arthritis, Vergrößerung sein Lymphknoten, Gelenkentzündungen, Heiserkeit und Knoten unter der Haut, die sich im weiteren Verlauf um die Gelenke verdicken Krankheiten.
Betroffene Personen mit Atembeschwerden müssen eventuell einen Atemschlauch einführen. Die meisten Kinder mit dieser Erkrankung sterben im Alter von 2 Jahren, normalerweise an Lungenkrankheit. Bei einer der schwersten Formen der Erkrankung kann kurz nach der Geburt eine vergrößerte Leber und Milz diagnostiziert werden. Babys, die mit dieser Form der Krankheit geboren wurden, sterben normalerweise innerhalb von 6 Monaten.
Morbus Farber wird durch einen Mangel an einem Enzym namens Ceramidase verursacht. Derzeit gibt es keine spezifische Behandlung für Morbus Farber. Zur Schmerzlinderung können Kortikosteroide verschrieben werden. Eine Knochenmarktransplantation kann Granulome (kleine Ansammlungen von entzündetem Gewebe) bei Patienten mit leichter Entzündung oder bei Patienten ohne Komplikationen des Lungen- oder Nervensystems reduzieren. Bei älteren Menschen können Granulome verkleinert oder operativ entfernt werden.
Gangliosidose besteht aus zwei verschiedenen Gruppen von genetischen Erkrankungen. Beide sind autosomal-rezessiv und betreffen Männer und Frauen gleichermaßen.
GM1-Gangliosidose.
Die GM1-Gangliosidose wird durch einen Mangel des Enzyms Beta-Galactosidase verursacht, was zu einer abnormalen Akkumulation von saure Lipidmaterialien, insbesondere in Nervenzellen des zentralen und peripheren Nervensystems Systeme. Die GM1-Gangliosidose hat drei klinische Manifestationen:
- GM1 (der schwerste Subtyp, der kurz nach der Geburt auftritt) kann Neurodegeneration, Krampfanfälle, vergrößerte Leber und Milz, Grobheit der Gesichtszüge, Skelettunregelmäßigkeiten, Gelenksteifheit, Blähungen, Muskelschwäche, übertriebene Schreckreaktion und Probleme mit Gangart. Etwa die Hälfte der Betroffenen entwickelt kirschrote Flecken im Auge. Kinder können im Alter von 1 Jahr taub und blind sein und sterben oft im Alter von 3 Jahren an Herzkomplikationen oder Lungenentzündung.
- Spätes Kind Die GM1-Gangliosidose beginnt normalerweise im Alter zwischen 1 und 3 Jahren. Neurologische Symptome sind Ataxie, Krampfanfälle, Demenz und Schwierigkeiten beim Sprechen.
- GM1-Gangliosidose entwickelt sich im Alter zwischen 3 und 30 Jahren. Zu den Symptomen gehören eine verminderte Muskelmasse (Muskelatrophie), neurologische Komplikationen, die weniger schwerwiegend sind und langsamer fortschreiten als bei anderen Formen Hornhauttrübung bei manchen Menschen und anhaltende Muskelkontraktionen, die zu Verdrehungen und sich wiederholenden Bewegungen oder abnormalen Körperhaltungen führen (Dystonie). Angiokeratome können sich im unteren Rumpf des Körpers entwickeln. Die Größe von Leber und Milz ist bei den meisten Betroffenen normal.
GM2-Gangliosidose.
Die GM2-Gangliosidose führt auch dazu, dass der Körper überschüssige saure Fettstoffe in Geweben und Zellen, vor allem in Nervenzellen, ansammelt. Diese Störungen resultieren aus einem Mangel des Enzyms Beta-Hexosaminidase. GM2-Störungen umfassen:
- Tay-Sachs-Krankheit (auch bekannt als GM2-Gangliosidose Variante B) und ihre Varianten werden durch einen Mangel des Enzyms Hexosaminidase A verursacht. Betroffene Kinder entwickeln sich in den ersten Lebensmonaten normal. Die Symptome beginnen im Alter von 6 Monaten und umfassen fortschreitenden Verlust der geistigen Leistungsfähigkeit, Demenz, verminderter Augenkontakt, erhöhtes eine erschreckende Reaktion auf Lärm, fortschreitender Hörverlust, der zu Taubheit führt, Schluckbeschwerden, Blindheit, kirschrote Flecken auf der Netzhaut und einige Lähmung. Anfälle können im zweiten Lebensjahr eines Kindes beginnen. Babys können schließlich von der Ernährungssonde abhängig werden und sterben oft im Alter von 4 Jahren an wiederkehrenden Infektionen. Leider gibt es keine Sonderbehandlung. Antikonvulsiva können anfänglich Anfälle kontrollieren. Andere unterstützende Behandlungen umfassen die richtige Ernährung und Flüssigkeitszufuhr sowie Methoden zum Offenhalten der Atemwege. Eine seltene Form der Erkrankung namens spät einsetzende Tay-Sachs-Krankheit, tritt bei Menschen im Alter zwischen 20 und 30 Jahren auf und ist durch einen instabilen Gang und eine fortschreitende neurologische Verschlechterung gekennzeichnet.
- Morbus Sandhoff (Option AB) ist eine schwere Form der Tay-Sachs-Krankheit. Der Beginn tritt normalerweise im Alter von 6 Monaten auf und ist nicht auf eine ethnische Gruppe beschränkt. Neurologische Anzeichen können eine fortschreitende Verschlechterung des Zentralnervensystems, der motorischen Schwäche, frühe Blindheit, starke Angstreaktion auf Geräusche, Spastik, Schock oder Zucken Muskeln (Myoklonus), Epilepsie, ein abnormal vergrößerter Kopf (Makrozephalie) und rote Flecken im Auge. Andere Symptome können häufige Atemwegsinfektionen, Herzgeräusch, puppenartige Gesichtszüge und Vergrößerung von Leber und Milz. Es gibt keine spezifische Behandlung für Morbus Sandhoff. Wie bei der Tay-Sachs-Krankheit umfasst die unterstützende Behandlung das Offenhalten der Atemwege sowie die richtige Ernährung und Flüssigkeitszufuhr. Antikonvulsiva können anfänglich Epilepsie (Krampfanfälle) kontrollieren.
Krabbes Krankheit(auch bekannt als globuläre Zellleukodystrophie und Galactosylceramid-Lipidose) ist eine autosomal-rezessiv vererbte Erkrankung, die durch einen Mangel des Enzyms Galactocerebrosidase verursacht wird. Die Krankheit betrifft am häufigsten Säuglinge mit Beginn vor dem 6. Lebensmonat, kann aber auch im Jugend- oder Erwachsenenalter auftreten.
Die Ansammlung von unverdauten Fetten beeinträchtigt das Wachstum der Nervenschutzhülle (Myelinscheide) und führt zu schweren Beeinträchtigungen der geistigen und motorischen Fähigkeiten. Andere Symptome sind Muskelschwäche, verminderte Dehnungsfähigkeit der Muskeln (Hypotonie), Muskelspannung (Spastizität), plötzlicher Schock oder Zucken der Gliedmaßen (myoklonische Anfälle), Reizbarkeit, unerklärliches Fieber, Taubheit, Blindheit, Lähmung und Schluckbeschwerden. Es kann auch zu einem langfristigen Gewichtsverlust kommen.
Die Krankheit kann durch Enzymtests und Identifizierung ihrer charakteristischen Zellgruppierung diagnostiziert werden in globoide Körper in der weißen Substanz des Gehirns, Demyelinisierung von Nerven und Degeneration und Zerstörung von Gehirnzellen. Bei Säuglingen verläuft die Krankheit in der Regel vor dem 2. Lebensjahr tödlich. Menschen mit einer fortgeschritteneren Form der Erkrankung haben einen milderen Krankheitsverlauf und leben deutlich länger.
Es wurde keine spezifische Behandlung für die Krabbe-Krankheit entwickelt, obwohl eine frühzeitige Knochenmarktransplantation einigen Menschen helfen kann. Menschen mit einer fortgeschritteneren Form der Erkrankung haben einen milderen Krankheitsverlauf und leben deutlich länger.
Metachromatische Leukodystrophie, oder MLD, ist eine Gruppe von Erkrankungen, die durch die Ansammlung von Fettstoffen in der weißen Substanz des Zentralnervensystems und in peripheren Nerven und teilweise in den Nieren gekennzeichnet sind. Ähnlich wie bei der Krabben-Krankheit betrifft MLD das Myelin, das die Nerven bedeckt und schützt. Diese autosomal-rezessiv vererbte Erkrankung wird durch einen Mangel des Enzyms Arylsulfatase A verursacht. Diese Störung betrifft sowohl Männer als auch Frauen.
Die metachromatische Leukodystrophie hat drei charakteristische Formen: spätes Kind, Jugendliche und Erwachsene.
- Späte Säuglings-MLD beginnt in der Regel zwischen 12 und 20 Monaten nach der Geburt. Kinder können zunächst normal erscheinen, haben aber Schwierigkeiten beim Gehen und neigen zum Fallen, begleitet von intermittierenden Schmerzen in Armen und Beinen. fortschreitender Sehverlust, der zu Blindheit, Entwicklungsverzögerungen und Verlust zuvor erworbener Meilensteine, Schluckstörungen, Krampfanfällen und Demenz bis zu 2. führt Jahre. Kinder entwickeln auch progressiven Muskelschwund und Schwäche und verlieren schließlich ihre Fähigkeit zu gehen. Die meisten Kinder mit dieser Form der Erkrankung sterben im Alter von 5 Jahren.
- Jugendliche MLD beginnt in der Regel im Alter zwischen 3 und 10 Jahren. Zu den Symptomen gehören eine eingeschränkte Schulleistung, geistige Beeinträchtigung, Ataxie, Krampfanfälle und Demenz. Die Symptome schreiten fort, wobei der Tod 10–20 Jahre nach Ausbruch der Krankheit eintritt.
- Erwachsenenform. Symptome Erwachsene Beginn nach dem 16. Lebensjahr und kann Ataxie, Krampfanfälle, abnormales Zittern der Gliedmaßen (Tremor), Konzentrationsstörungen, Depression, psychische Störungen und Demenz. Der Tod tritt in der Regel 6–14 Jahre nach Einsetzen der Symptome ein.
MLD kann nicht geheilt werden. Die Behandlung ist symptomatisch und unterstützend. Eine Knochenmarktransplantation kann in einigen Fällen das Fortschreiten der Erkrankung verzögern. Signifikante Fortschritte wurden bei der Gentherapie in Tiermodellen mit MLD und in klinischen Studien erzielt.
Wolman-Krankheitauch bekannt als Mangel an lysosomaler saurer Lipaseist eine schwere Lipidspeicherstörung, die im Alter von 1 Jahr normalerweise tödlich verläuft. Diese autosomal-rezessiv vererbte Erkrankung ist gekennzeichnet durch eine Akkumulation von Cholesterinestern (normalerweise die Transportform von Cholesterin) und Triglyceride (die chemische Form, in der Fette im Körper vorkommen), die sich erheblich ansammeln und Zellschäden verursachen können und Stoffe.
Sowohl Männer als auch Frauen leiden an dieser Störung. Säuglinge sind bei der Geburt normal und aktiv, entwickeln jedoch schnell fortschreitende geistige Beeinträchtigungen, eine vergrößerte Leber und stark vergrößerte Milz, Blähungen, Magen-Darm-Probleme. Gelbsucht, Anämie, Erbrechen und Kalkablagerungen in Nebennierenwodurch sie hart werden.
Ein anderer Typ Ein Mangel an lysosomaler saurer Lipase ist Cholesterinester-Speicherkrankheit. Diese extrem seltene Krankheit entsteht durch die Einlagerung von Cholesterinestern und Triglyceride in Blutzellen, Lymphe und Lymphgewebe. Bei Kindern im Erwachsenenalter vergrößert sich die Leber, was zu Zirrhose und chronisch Leberversagen. Kinder können auch Kalziumablagerungen in den Nebennieren haben, und in den späteren Stadien der Krankheit kann sich Gelbsucht entwickeln.
Derzeit wird die Frage des Enzymersatzes sowohl bei der Wolman-Krankheit als auch bei der Akkumulation eines Cholesterinesters aktiv untersucht.
Beachtung! Die Informationen dienen nur zu Informationszwecken und dienen nicht der Diagnose und Verschreibung einer Behandlung. Konsultieren Sie immer Ihren Facharzt!