Phenylketonurie
 Phenylketonurie( Felling-Krankheit) - eine schwere Erbkrankheit, die durch Stoffwechselstörungen von Aminosäuren und wird durch fortschreitende Demenz, Verzögerung der körperlichen Entwicklung, Muskeltonus Störungen und Bewegungen manifestiert. Diese Pathologie tritt in 1 von 10.000 Neugeborenen. Mit dem Geschlechterverhältnis ist das gleiche, aber die meisten Jungen mit Phenylketonurie während des ersten Lebensjahres sterben.
Phenylketonurie( Felling-Krankheit) - eine schwere Erbkrankheit, die durch Stoffwechselstörungen von Aminosäuren und wird durch fortschreitende Demenz, Verzögerung der körperlichen Entwicklung, Muskeltonus Störungen und Bewegungen manifestiert. Diese Pathologie tritt in 1 von 10.000 Neugeborenen. Mit dem Geschlechterverhältnis ist das gleiche, aber die meisten Jungen mit Phenylketonurie während des ersten Lebensjahres sterben.
Basis von Phenylketonurie in Synthese ist unzureichend Umwandlung Phenylalanin Enzym Phenylalanin-4-hydroxylase Tyrosin. Als Folge dieses Mangels in den Flüssigkeiten / Gewebe des Körpers ist eine signifikante Häufung von Phenylalanin und seine Derivate( feniletillamin, fenilatsetilglyutamin und Phenylmilchsäure, Phenylbrenztraubensäure und Phenylessigsäure), die gegenüber den toxischen Läsionen des zentralen Nervensystems führt, wodurch Störungen im Stoffwechsel der Hormone sind, das ProteinAustausch;mit Aufschlüsselung des Transports von Aminosäuren Austausch von Serotonin und Katecholamine
Phenylketonurie gestört ist -
Symptome in der ersten Zeit nach der Geburt, sieht das Baby gesund. Am häufigsten ist die ersten Symptome von Phenylketonurie im Altersbereich von zwei bis sechs Monaten, es ist: der Mangel an Interesse in jeder Umgebung, Lethargie markiert oder entgegengesetzte Reizbarkeit, Erbrechen, Unruhe. Da das Kind Überwindung sechs Monate alt ist, wird eine deutliche Verzögerung bei der geistigen Entwicklung: 60% der Kinder ein Idiot ist, 10% der Kinder - Phenylbrenztraubensäure mentale Retardierung .Der Schädel ist in seiner Größe leicht verringert, das Wachstum kann entweder normal oder leicht reduziert, beobachtet später Zahnen Kinder zu spät zu lernen, zu sitzen und zu gehen. Gang und Haltung in diesen Kindern ganz eigentümlich: sie stehen mit gespreizten Beinen, an den Hüften und Knie zu beugen, hängende Schultern und Kopf;geht schwankend in kleinen Schritten. Aufgrund des erhöhten Muskeltonus, sitzen solche Kinder mit gekreuzten Beinen( Schneider Pose).In den meisten Fällen Kleinkinder blauäugig, blond, Haut fast völlig frei von Pigment. Einige Patienten haben Alters Anfälle vorbei.
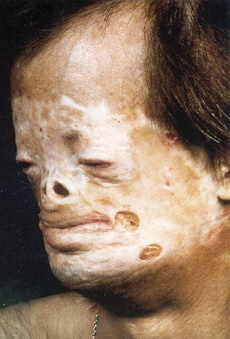
charakteristischsten Manifestationen von Phenylketonurie: geistige Behinderung, psychische und neurologische Störungen, Angst( als Kind), eine spezifische Haltung und Haltung beim Sitzen, eigentümlicher Gang, klischee Bewegungen, ungewöhnliche Position der Gliedmaßen, Krämpfe, erhöhten Sehnenreflexe, fehlerhafte Ausbildung der Myelin ÄnderungenHaut, Mikrocephalie , Hypopigmentierung, Hautveränderungen, Ekzeme, Trockenheit, Katarakt, Licht iris, Sklerodermie, eine deutliche „mouseth ‚Körpergeruch, Erbrechen in der Neugeborenenperiode ist hypopigmentierte Haar
Diagnostics
Phenylketonurie sehr wichtig, eine Diagnose bereits in der präklinischen Phase, oder zumindest spätestens im zweiten Monat des Babys das Leben, da das mögliche Auftreten der ersten Anzeichen der Krankheit zu etablieren. Alle Neugeborenen unterliegen der Kontrolle durch eine spezielle Screening-Programme, die zum Nachweis einer erhöhten Konzentration von Phenylalanin im Blut der Lage sind, auch in den ersten Lebenswochen. Jedes Kind mit den erfassten minimalen neurologischen Symptome oder Anzeichen einer Entwicklungsverzögerung sollte auf das Vorhandensein von Phenylalanin Stoffwechsel Pathologie untersucht werden. Differentialdiagnostisch intrauterine Infektionen und intrakraniellen Geburtstrauma
Phenylketonurie - Behandlung
einzig wirksame Behandlung für PKU gilt seit den ersten Tagen des Lebens organisiert werden speziell Diät-Therapie entwickelt, deren Prinzip ist in Phenylalanin Lebensmittel enthalten sind, zu begrenzen, die solche Lebensmittel wie Brot ausgeschlossenProdukte, Nüsse, Schokolade, Getreide, Hülsenfrüchte, Eier, Käse, Fisch, Fleisch und so weiter. Die therapeutische Diät Patienten fenilketonuriey besteht aus spezialisierten Produkten, sowohl ausländische als auch inländische Produktion. Kinder ersten Lebensjahres zeigen die Produkte in ihrer Zusammensetzung in der Nähe der Muttermilch, es eine solche Mischung als „Lofenilak“ und „Afenilak“ ist. Für Kinder, die älter ein wenig solche Mischungen als "Tetrafen", "Maksamum-XP", "Phenyl-Free" entwickelt. PKU schwangere Frauen und ältere Kinder( nach sechs Jahren) zeigen eine typische Mischung „Maksamum-XP“.Zusätzlich zu den spezialisierten Medizinprodukten in der Ernährung des Patienten gehören Säfte, Obst und Gemüse.
unter Behandlung Kinder müssen unter der ständigen Aufsicht eines Kinderarzt und Neuropsychiater sein. Zu Beginn der Behandlung von Phenylketonurie Phenylalaningehalts Kontrolle wird auf wöchentlicher Basis durchgeführt, mit der Normalisierung des Schalters 1 mal pro Monat während des ersten Lebensjahres und 1 Mal in zwei Monaten in Kindern, die älter als ein Jahr.
Zeit begonnen Diät-Therapie verhindert oft die Entwicklung von charakteristischen klinischen Manifestationen der klassischen Phenylketonurie. Die Behandlung wird vor der Pubertät ohne Fehler durchgeführt, und manchmal auch länger. Da Patienten mit Phenylketonurie Frau eine gesunde Fötus machen nicht in der Lage ist, gehalten, auch vor der Empfängnis begonnen und bis zur Geburt eine besondere Behandlung unter Ausschluss des fetalen Phenylalanin aus
kranke Mutter Weitere Artikel zum Thema Weiterbildung up:
1. Tay-Sachs-4. Marfan Syndrom
2. 5. Klinefelter-Syndrom Patau Syndrom
3. Edwards-Syndrom Syndrom 6. Lejeune