Gaucherin tauti
 Gaucherin tauti on perinnöllinen kertyvä sairaus, joka on yksi yleisimmistä lysosomaalisten patologioiden joukossa. Gaucherin tauti kehittyy johtuen glukoserebrosidaasin riittämättömästä vaikutuksesta, joka johtuu glukoserebrosidin kertymisestä kudoksissa ja eräissä elimissä.
Gaucherin tauti on perinnöllinen kertyvä sairaus, joka on yksi yleisimmistä lysosomaalisten patologioiden joukossa. Gaucherin tauti kehittyy johtuen glukoserebrosidaasin riittämättömästä vaikutuksesta, joka johtuu glukoserebrosidin kertymisestä kudoksissa ja eräissä elimissä.
Tämä tauti on suhteellisen harvinaista tautia ja ilmenee tilastojen mukaan suhteellisen alhaisena. Mutta se voi kuitenkin johtaa vammaisuuteen ja / tai kuolemaan varhaisessa iässä, jos asianmukaista hoitoa ei aloiteta ajoissa.
Gaucherin tauti aiheuttaa
: tä Geneettisesti mutaatiot esiintyvät geeneissä, jotka ovat vastuussa glukooserebrosidaasientsyymin tuottamisesta. Tämä geeni poikkeavuuksilla on lokalisoitu kromosomin 1-kromosomiin. Nämä mutaatiot aiheuttavat entsyymin vähäisen aktiivisuuden. Siten glukooserebrosidien kertyminen makrofageissa on.
Meshchymal-solut, joita kutsutaan Gaucher-soluiksi, laajenevat asteittain ja hypertrofiavat. Kun nämä solut muuttuvat, ja ne ovat perna, munuaiset, maksa, keuhkot, pää ja luuytimessä, ne puolestaan vääristävät näitä elimiä ja häiritsevät niiden normaalia toimintaa.
Gaucherin tauti viittaa autosomaalisiin resessiivisiin sairauksiin. Siksi jokainen henkilö voi periä tämän entsyymin mutaation kaikilla ominaisuuksilla samassa suhteessa sekä isältä että äidiltä.Siten taudin aste ja sen vakavuus riippuvat geenien vaurioitumisesta.
Teoriassa kukin henkilö voi periä glukoserebrosidigeenin, jossa on vaurioita tai täysin terveitä.Anomalioiden omaavan geenin perimän seurauksena esiintyy tämän entsyymin mutaatio, mutta tämä ei puhu taudista. Mutta kun lapsi saa molemmat sairaudet, Gaucherin tauti diagnosoidaan. Jos jompikumpi kyseessä olevista geeneistä on peritty, lapsen katsotaan olevan taudin kantaja, joten on todennäköistä, että tämä ominaisuus ja perinnöllinen patologia tulevat tuleville sukupolville. Näin ollen lapsen syntyminen Gaucherin taudin ollessa kyseessä kahdessa vanhemmassa voi tapahtua, kun lapsi on syntynyt 25 prosentissa tapauksista, lapsen kuljettaja 50 prosenttia ja terve lapsi 25 prosentilla.
Tämän perinnöllisen patologian esiintyvyys etnisten rotujen kesken on 1: 50.000, mutta sitä esiintyy useammin Ashkenazi-juutalaisten keskuudessa.
Gaucherin tautia kutsutaan myös kerääntymisen sairaudeksi riittämättömän entsyymin takia, jonka on poistettava haitalliset aineenvaihduntatuotteet kehosta eikä kerätä niitä.Tämän seurauksena nämä aineet kerääntyvät eräiden elinten makrofageihin ja tuhoavat ne.
Gaucherin tauti
-oireet Taudin kliinistä kuvaa on tyypiltään kolme tyyppiä.
Ensimmäisessä Gaucher-tautityypissä hermosto ei osallistu lipidien kerääntymiseen, joten se viitataan enemmän taudin hematologiseen kulkuun. Ensinnäkin monissa parenkymaalisissa elimissä on kasvua, joista ensimmäinen on perna ja sitten havaitaan luiden visuaaliset muodonmuutokset. Paljon usein tällainen sairaus etenee aikuisten keskuudessa.
Infant tai toisen tyypin Gaucher-tauti on luonteenomaista neurologian akuutti muoto ja diagnosoidaan pääasiassa jopa kuuden kuukauden ikäisenä.Tässä tapauksessa hyvin tyypilliset neurologiset oireet ilmestyvät hyvin varhaisessa vaiheessa, jolloin aivokudosolut ovat vaurioituneita, mikä aiheuttaa lasten kuoleman.
Kolmas Gaucher-tautityyppi on sairauden nuorten muoto, jolla on subakuutti neurovisaalinen tyyppi. Patologisen prosessin alussa muodostuu splenomegalia ja henkinen hidastuminen, jossa on vaikutusta pyramidaalisiin ja ekstrapyramidaalisiin aivojärjestelmiin.
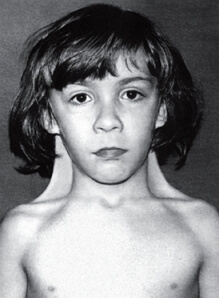
Gaucher -taudilla on merkittäviä muutoksia luuytimen, pernan, imusolmukkeiden ja maksan vaurioitumisen muodossa, aivojen vaikutus aivojen muotoon. Parenkymaalisissa elimissä on suuri Gaucher-solujen pitoisuus, ja nykyisten nekroosialueiden kanssa aivokuoressa esiintyy hajakuormittavia muutoksia. Tutkittaessa potilaita merkitsi jyrkästi suurentunutta pernaa ja maksa, ja joskus imusolmukkeita. Kehossa esiintyy ominaispiirteitä tälle tummansiniselle värisävylle.
Gaucherin tauti on tunnusomainen oire taudin klinikalla lisäyksenä pernassa alussa kehittää patologian, ja sitten on koon kasvu ja maksassa. Kasvoille, käsille, selkäreille ja limakalvoille havaitaan tietty pigmentti. Lipidi kertymistä luuytimessä johtaa osteoporoosiin, nikamien, reisiluu, ja spontaaneja murtumia, anemia, trombosytopenia ja leukopenia. Joskus vastasyntyneillä on jopa pahanlaatuinen sairaus, joka esiintyy spastisen oireyhtymän, strabismuksen ja pertussis-kaltaisen yskän muodossa.
Gaucherin tautitapauksissa havaitaan hepatosplenomegalia suurentuneiden viskeraalisten imusolmukkeiden kanssa. Kehon avoimet osat pigmentoituvat kellertävän ruskealla värillä, joka myöhemmin muuttuu pronssiksi johtuen tietyn pigmentin levittämisestä ihon alle. Potilaat valittavat yleensä vatsakipu, tyypilliset epämuodostumat ja usein luiden murtumat. Edistyneessä iässä on hyvin määriteltyjä verisuonten aterooma-oireita samanaikaisesti kohonneen kolesterolemian kanssa.
Gaucherin taudin viimeisessä vaiheessa vaikeat neurologiset häiriöt kehittyvät hyvin usein, mikä johtaa kuolemaan.
Gaucherin taudin diagnoosi
Nykyään käytetään erilaisia menetelmiä Gaucherin taudin diagnosoimiseksi.
Yksi tarkimmista on verikokeessa, jossa määritetään entsyymin läsnäolo ja glukoserebrosidaasin taso leukosyyteissä.
DNA-analyysissä tarkastellaan toista menetelmää, geneettisellä tasolla olevia mutaatioita ja emäksisen entsyymin puuttuvaa määrää.Tämä menetelmä perustuu viimeisimpään molekyylibiologian teknologiaan. Gaucherin taudin diagnoosin tärkein etu on se, että se mahdollistaa raskauden alkuvaiheen tutkimukset. Ja tämä sallii 90% tulevaisuudessa määrittää taudin harjoittajan ja jopa sen vakavuuden.
Kolmannessa diagnoosimenetelmässä tutkitaan luuydintä, jonka ansiosta sen solujen toimintahäiriöt voidaan määrittää.Tällaisen tutkimuksen kielteinen puoli on, että se sallii vain potilaiden diagnosoinnin, mutta se ei pysty tunnistamaan taudin kantajia. Siksi nykyään tällaista menetelmää ei käytännössä sovelleta.
Valitettavasti joskus Gaucherin taudin oireet eivät aina ole helppoa. Siksi oikea ja oikea-aikainen diagnoosi voi pidentää potilaan elämää jo vuosia. Tällöin potilas on asiantuntijoiden( pediatrian ja hematologin) valvonnassa ilman hoitoa ja ilman pelkoa komplikaatioista. Toisaalta, jos potilas ei ole läpäissyt ajoissa pätevää diagnoosia ja ei saanut lääketieteellistä apua, voi elinikäinen saada taudin oireita, kärsivät niistä eikä epäillä, että hänellä oli Gaucherin tauti, vastaavasti, ja ei saanut asianmukaista hoitoa.
Gaucherin taudinkäsittely
Viime aikoihin saakka tautia hoidettiin sen oireiden mukaan: perna poistettiin tai toimenpiteet suoritettiin patologisissa murtumissa. Mutta vuonna 1991 USA: ssa ilmestyi ensimmäinen lääketieteellinen valmiste glukaraasi, joka mahdollistaa Gaucherin taudin hoitamisen. Tämä tauti oli ensimmäinen kertyvyyssairauksien joukossa, joka oli herkkä entsyymikorvaushoidolle.
Gaucherin taudin entsyymikorvaushoidon toinen tuote, imigluseraasi, hyväksyttiin virallisesti vuonna 1994.Nämä lääkkeet ovat ihmisen entsyymi-glukoserebrosidaasin analogeja, jotka on tuotettu viimeisimmän tekniikan avulla DNA: n keinotekoiseksi tekemiseksi.
Tällä hetkellä suuri määrä Gaucherin tautia sairastavilla potilailla ympäri maailmaa vastaanottaa jatkuvasti entsyymikorvaushoitoa muunnettujen beeta-glukoserebrosidaasin muotojen muodossa. Näitä ovat ceradaasi( alglukeraasi) tai ceresiimi( imigluseraasi injektioissa).Nämä lääkkeet on suunniteltu siten, että ne voivat spesifisesti kohdistaa infektioita tuhoavia valkosoluja( makrofageja) nopeuttaakseen glukoserebrosidien jakautumista glukoosiksi ja ceramidiksi.
Osoitti Gaucherin taudin hoidon menestyksen cerevisiaa käyttäen 60 yksikköä / kg kohdalla, joka levitettiin joka toinen viikko. Kurssin rekisteröity mitä tämä annos vähentää merkittävästi organomegalian, vähentää usein sisäelimissä koon ja komplikaatio hematologisten etiologies sekä parantaa potilaiden elämää ensimmäisen tyypin Gaucherin tautia.
Tätä patologista perinnöllisyyttä on vuodesta 1997 lähtien käsitelty entsyymikorvaushoidolla Venäjällä.Ensimmäisen tyypin potilaille Cereziman nimittäminen alkaa 30 yksikköä kilogrammalta kerran päivässä.Kuusi kuukautta Gaucherin taudin hoidon aloittamisen jälkeen havaitaan positiivinen dynamiikka monissa parenchymaalisissa elimissä ja myös hematologisissa indekseissä.
Gaucherin taudin pitkäaikaishoito tällä lääkkeellä täysin vakauttaa patologisen prosessin, vähentää huomattavia muutoksia luissa ja parantaa huomattavasti potilaiden elämää.Siksi aikaisempi vastaava hoito aloitetaan, sitä tehokkaammat tulokset ovat.
Tärkeä näkökohta on Gauchersin taudin ehkäiseminen, joka käsittää perheiden lääketieteellisen geneettisen neuvonnan ja synnytyksen diagnoosin. Tämä mahdollistaa glukooserebrosidaasin aktiivisuuden määrittämisen sikiön verhokäyrän ja sen napanuoran verisolujen alkuvaiheissa.