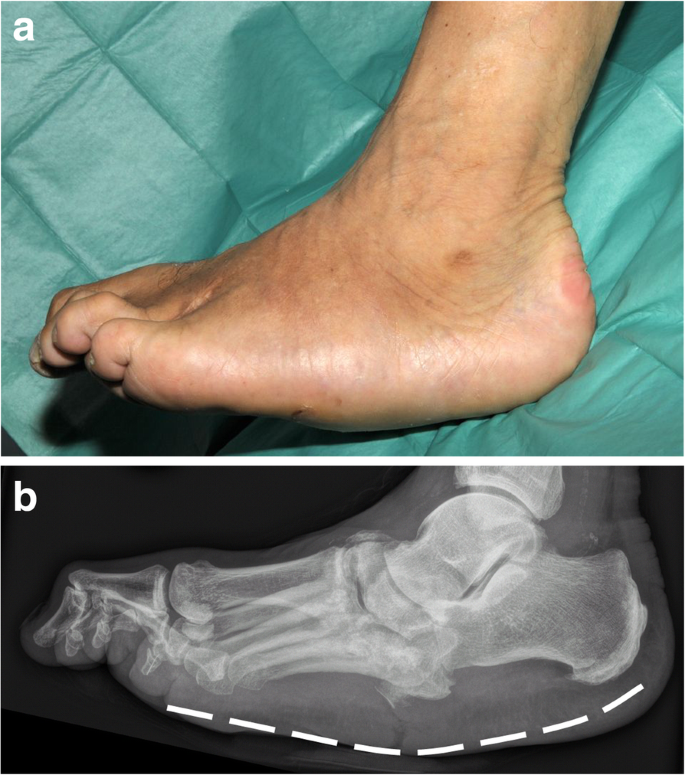
Lipoïdose (maladie de stockage des lipides lysosomal): qu'est-ce que c'est, exemples
Attention! Les informations sont à titre informatif uniquement et ne sont pas destinées à diagnostiquer et à prescrire un traitement. Consultez toujours votre médecin spécialiste !
Teneur
- Que sont les maladies de surcharge lysosomale ?
- Que sont les lipides ?
- Comment se transmet la lipoïdose ?
- Types et symptômes des maladies de surcharge lysosomale
Que sont les maladies de surcharge lysosomale ?
Maladies de surcharge lysosomale (lipoïdose), sont un groupe de maladies métaboliques héréditaires dans lesquelles des quantités nocives de matières grasses (lipides) s'accumulent dans diverses cellules et tissus du corps.
Les personnes atteintes de ces troubles ne fabriquent pas suffisamment l'une des enzymes nécessaires pour décomposer (métaboliser) les lipides, ou elles produisent des enzymes qui ne fonctionnent pas correctement.
Au fil du temps, cette accumulation excessive de graisse peut entraîner des dommages permanents aux cellules et aux tissus, en particulier dans le cerveau, le système nerveux périphérique (nerfs de la moelle épinière au reste de la corps),
le foie, rate et la moelle osseuse.Que sont les lipides ?
Les lipides sont des substances semblables à de la graisse qui sont des parties importantes des membranes à l'intérieur et entre les cellules et dans la gaine de myéline, qui recouvre et protège les nerfs. Les lipides comprennent les huiles, les acides gras, les cires, les stéroïdes (tels que le cholestérol et les œstrogènes) et d'autres composés apparentés.
Ces corps gras sont naturellement stockés dans les cellules, les organes et les tissus de l'organisme. De minuscules corps dans les cellules, appelés lysosomes, convertissent ou métabolisent régulièrement les lipides et les protéines en composants plus petits pour fournir de l'énergie au corps. Les troubles dans lesquels le matériel intracellulaire, qui ne peut pas être métabolisé, reste dans les lysosomes, sont appelés maladies de surcharge lysosomale.
En plus des maladies associées à l'accumulation de lipides, d'autres maladies lysosomales associées à l'accumulation comprennent les mucolipidoses, dans lesquelles les cellules et les tissus s'accumulent. une quantité excessive de lipides auxquels sont attachées des molécules de sucre, ainsi que des mucopolysaccharidoses, dans lesquelles une quantité excessive de sucre complexe et volumineux s'accumule molécules.
Comment sont hérités lipoïdose?
Les maladies de surcharge lysosomale sont héritées d'un ou des deux parents qui portent un gène défectueux qui régule une enzyme spécifique de métabolisation des lipides dans une classe de cellules du corps. Ils peuvent être hérités de deux manières :
- Hérédité autosomique récessive se produit lorsque les deux parents portent et transmettent une copie du gène défectueux, mais aucun des parents n'est affecté par le trouble. Chaque enfant né de ces parents a 25 pour cent de chance d'hériter des deux copies du gène défectueux, 50 pour cent la probabilité qu'il soit un porteur similaire à ses parents, et une probabilité de 25 pour cent de n'hériter d'aucune copie du document défectueux gène. Les enfants des deux sexes peuvent être hérités de manière autosomique récessive.
- Hérédité récessive liée à l'X (ou liée au sexe) se produit lorsque la mère porte le gène affecté sur le chromosome X. Les chromosomes X et Y sont impliqués dans la détermination du sexe. Les femmes ont deux chromosomes X, tandis que les hommes ont un chromosome X et un chromosome Y. Les fils de femmes porteuses ont 50% de chances d'hériter et d'affecter la maladie, car les fils reçoivent un chromosome X de leur mère et un chromosome Y de leur père. Les filles ont 50% de chances d'hériter du chromosome X affecté de leur mère et sont porteuses ou légèrement affectées. Les hommes atteints ne transmettent pas la maladie à leurs fils, mais leurs filles seront porteuses et porteuses de la maladie.
Types et symptômes des maladies de surcharge lysosomale
maladie de Gaucher - la plus fréquente des maladies lysosomales. La maladie est causée par une déficience de l'enzyme glucocérébrosidase, qui conduit à l'accumulation de glucocérébroside dans de nombreux tissus. Les matières grasses peuvent s'accumuler dans le cerveau, la rate, le foie, les reins, les poumons et la moelle osseuse.
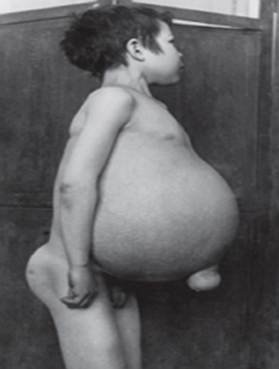
Les symptômes peuvent inclure des lésions cérébrales, hypertrophie de la rate et du foie, altération de la fonction hépatique, troubles squelettiques et lésions osseuses pouvant provoquer des douleurs et des fractures, gonflement des ganglions lymphatiques et (parfois) des articulations adjacentes, ballonnement, teint brunâtre, anémie, une faible numération plaquettaire et des taches jaunes sur les yeux.
Les personnes les plus gravement touchées peuvent également être plus sensibles aux infections. La maladie touche autant les hommes que les femmes.
La maladie de Gaucher comporte trois sous-types cliniques généraux :
- Type 1 (Ou pas neuronopathique type) est la forme la plus courante de la maladie aux États-Unis et en Europe. Le cerveau n'est pas affecté, mais il peut y avoir des dommages aux poumons et, dans de rares cas, aux reins. Les symptômes peuvent commencer tôt dans la vie ou à l'âge adulte et inclure une hypertrophie du foie et une hypertrophie sévère de la rate, ce qui peut entraîner une rupture et des complications supplémentaires. La faiblesse du squelette et la maladie osseuse peuvent être étendues. Les personnes de ce groupe ont généralement facilement des ecchymoses en raison de la faible numération plaquettaire dans leur sang. Ils peuvent également ressentir de la fatigue due à l'anémie. Selon l'apparition et la gravité de la maladie, les personnes atteintes de type 1 peut vivre jusqu'à l'âge adulte. De nombreuses personnes atteintes ont une maladie bénigne ou peuvent ne présenter aucun symptôme. Bien que 1taper La maladie de Gaucher est courante chez les personnes d'origine juive ashkénaze et peut toucher des personnes de toutes origines ethniques.
- Type 2 (ou neuropathique aigu de l'enfance maladie de Gaucher) débute généralement dans les 3 mois suivant la naissance. Les symptômes comprennent des lésions cérébrales étendues et progressives, une spasticité, des convulsions, des membres raides, une hypertrophie du foie (hépatomégalie hépatique) et la rate (splénomégalie), mouvements oculaires anormaux et faible capacité à sucer et à avaler. Les enfants atteints meurent généralement avant l'âge de 2 ans.
- Type 3 (neuronopathique chronique forme) peut commencer à tout moment pendant l'enfance ou même à l'âge adulte. Elle se caractérise par des symptômes neurologiques lentement progressifs mais moins sévères par rapport à la maladie de Gaucher aiguë 2 types. Les principaux symptômes comprennent des troubles du mouvement oculaire, des déficits cognitifs, une mauvaise coordination, des convulsions, hypertrophie de la rate et/ou du foie, troubles squelettiques, troubles sanguins, y compris l'anémie, et problèmes de respiration. Presque tout le monde avec la maladie de Gaucher 3 sortes, qui reçoit un traitement enzymatique substitutif atteindra l'âge adulte.
Pour les patients 1er et 3e types La thérapie de remplacement enzymatique, administrée par voie intraveineuse toutes les deux semaines, peut réduire considérablement la taille du foie et de la rate, réduire les anomalies squelettiques et inverser d'autres manifestations. Une greffe de moelle osseuse réussie traite les manifestations neurologiques de la maladie. Cependant, cette procédure comporte des risques importants et est rarement pratiquée chez les personnes atteintes de la maladie de Gaucher.
Dans de rares cas, une intervention chirurgicale peut être nécessaire pour retirer tout ou partie de la rate (si une personne a une très faible numération plaquettaire ou lorsqu'un organe hypertrophié affecte grandement le confort de la personne). La transfusion sanguine peut être bénéfique pour certaines personnes souffrant d'anémie. D'autres peuvent avoir besoin d'une chirurgie de remplacement articulaire pour améliorer la mobilité et la qualité de vie. Il n'existe actuellement aucun traitement efficace pour les lésions cérébrales qui peuvent survenir chez les personnes atteintes de la maladie de Gaucher de type 2 et 3.
Maladie de Niemann-Pick est un groupe de maladies autosomiques récessives causées par l'accumulation de graisse et de cholestérol dans les cellules du foie, de la rate, de la moelle osseuse, des poumons et, dans certains cas, du cerveau.
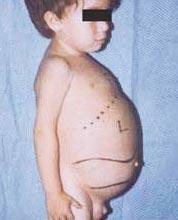
Les complications neurologiques peuvent inclure l'ataxie (manque de coordination musculaire pouvant affecter la marche, l'écriture et l'alimentation, entre autres fonctions), paralysie oculaire, dégénérescence cérébrale, problèmes d'apprentissage, spasticité, difficulté à s'alimenter et à avaler, parole, perte de tonus musculaire, hypersensibilité au toucher et une certaine opacité cornéenne due à une accumulation excessive matériaux. Chez 50% des personnes atteintes, un halo rouge cerise caractéristique se développe autour du centre de la rétine, qui peut être vu par un médecin avec un instrument spécial.
La maladie de Niemann-Pick est classée en trois sous-types généraux :
- Type A, la forme la plus sévère, commence dans la petite enfance. Les bébés semblent normaux à la naissance, mais à 6 mois, des lésions cérébrales profondes se développent, une hypertrophie du foie et de la rate, et des ganglions lymphatiques enflés et des ganglions sous la peau (xanthomes). La rate peut atteindre 10 fois sa taille normale et se rompre, provoquant des saignements. Ces enfants s'affaiblissent progressivement, perdent leur fonction motrice, peuvent devenir anémiques et sont sujets à des infections récurrentes. Ils vivent rarement plus de 18 mois. Cette forme de la maladie est la plus courante dans les familles juives.
- Type B(ou apparition juvénile) n'affecte généralement pas le cerveau, mais la plupart des enfants développent ataxie, lésion des nerfs quittant la moelle épinière (neuropathie périphérique), et des difficultés pulmonaires qui progressent avec l'âge. L'hypertrophie du foie et de la rate est typique chez les adolescents. Gens avec type B peuvent vivre relativement longtemps, mais beaucoup nécessitent une oxygénothérapie à vie en raison de lésions pulmonaires. Les types Niemann-Pick A et B sont le résultat d'une accumulation d'une substance grasse appelée sphingomyéline due à une déficience en une enzyme appelée sphingomyélinase.
- Type C peuvent apparaître tôt dans la vie ou se développer à l'adolescence ou même à l'âge adulte. Maladie de Niemann-Picktype C n'est pas causée par un déficit en sphlingomyélinase, mais par un manque de protéines NPC1 ou NPC2. De ce fait, divers lipides et notamment le cholestérol s'accumulent dans les cellules nerveuses et provoquent leur dysfonctionnement. L'atteinte cérébrale peut être étendue, entraînant une incapacité à regarder vers le haut et vers le bas, des difficultés à marcher et à avaler, une perte auditive progressive et démence. Gens avec type C n'ont qu'une légère hypertrophie de la rate et du foie. L'espérance de vie des personnes atteintes type C varie considérablement. Certaines personnes meurent dans l'enfance, tandis que d'autres, moins touchées, peuvent également vivre à l'âge adulte.
Il n'existe actuellement aucun remède contre la maladie de Niemann-Pick. Le traitement est de soutien. Les enfants meurent généralement d'une infection ou d'une perte neurologique progressive. Il y a eu des cas de greffe de moelle osseuse chez plusieurs patients atteints de type B avec des résultats mitigés.
La maladie de Fabryaussi connu sous le nom Déficit en alpha-galactosidase-A, provoque l'accumulation de matières grasses dans le système nerveux autonome (cette partie du système nerveux qui contrôle les fonctions involontaires telles que la respiration et le rythme cardiaque), dans les yeux, les reins et les maladies cardiovasculaires système.
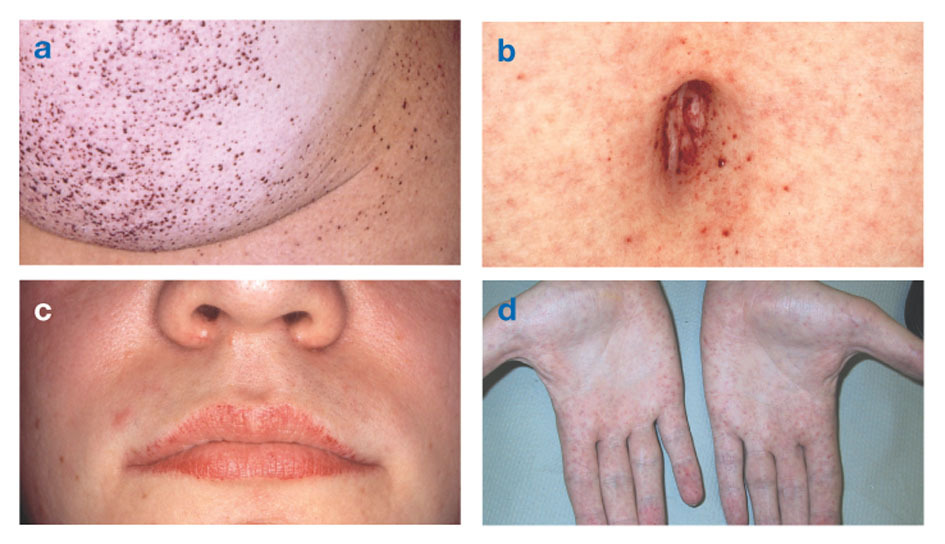
La maladie de Fabry est la seule maladie lipidique liée à l'X. La maladie affecte principalement les hommes, bien qu'une forme plus douce et plus variable se produise chez les femmes. Rarement, les femmes touchées présentent des symptômes graves similaires à ceux observés chez les hommes atteints de la maladie. L'apparition des symptômes survient généralement pendant l'enfance ou l'adolescence.
Les signes neurologiques comprennent des douleurs brûlantes dans les bras et les jambes qui s'aggravent par temps chaud ou après l'exercice, ainsi que l'accumulation de matière en excès dans les couches transparentes de la cornée (ce qui conduit à une opacification, mais sans changements de vision). L'accumulation de graisse dans les parois des vaisseaux sanguins peut perturber la circulation, mettant une personne en danger accident vasculaire cérébral ou crise cardiaque. D'autres symptômes incluent une hypertrophie du cœur, progressive insuffisance rénaleentraînant une insuffisance rénale terminale, des problèmes gastro-intestinaux, une diminution de la transpiration et de la fièvre.
Les angiokératomes (petites plaques surélevées, non cancéreuses, rouge-violet sur la peau) peuvent se développer dans le bas du torse et devenir plus fréquents avec l'âge.
Les personnes atteintes de la maladie de Fabry meurent souvent prématurément des complications de insuffisance cardiaque, insuffisance rénale ou accident vasculaire cérébral. Des médicaments comme la phénytoïne et la carbamazépine sont souvent prescrits pour traiter la douleur qui accompagne la maladie de Fabry, mais pas pour la guérir. Le métoclopramide ou le Lipisorb (complément alimentaire) peuvent soulager les troubles gastro-intestinaux qui survient souvent chez les personnes atteintes de la maladie de Fabry, et certaines personnes peuvent avoir besoin d'une greffe de rein ou dialyse. Le remplacement enzymatique peut réduire le stockage, soulager la douleur et préserver la fonction des organes chez certaines personnes atteintes de la maladie de Fabry.
La maladie de Farber aussi connu sous le nom La lipogranulomatose de Farber, décrit un groupe de maladies autosomiques récessives rares qui provoquent l'accumulation de matière grasse dans les articulations, les tissus et le système nerveux central. Elle touche aussi bien les hommes que les femmes. La maladie débute généralement dans la petite enfance, mais peut survenir plus tard dans la vie.
Les enfants atteints de la maladie de Farber classique se développent symptômes neurologiques, qui peuvent inclure une somnolence et une léthargie accrues, et des problèmes de avaler. Le foie, le cœur et les reins peuvent également être touchés. D'autres symptômes peuvent inclure des contractures articulaires (raccourcissement chronique des muscles ou des tendons autour des articulations), des vomissements, de l'arthrite, une hypertrophie ganglions lymphatiques, inflammation des articulations, enrouement et bosses sous la peau qui s'épaississent autour des articulations à mesure qu'elles progressent maladies.
Les personnes affectées ayant des difficultés respiratoires peuvent avoir besoin de se faire insérer un tube respiratoire. La plupart des enfants atteints de cette maladie meurent avant l'âge de 2 ans, généralement à partir de maladies pulmonaires. Dans l'une des formes les plus graves de la maladie, une hypertrophie du foie et de la rate peut être diagnostiquée peu de temps après la naissance. Les bébés nés avec cette forme de la maladie meurent généralement dans les 6 mois.
La maladie de Farber est causée par une déficience d'une enzyme appelée céramidase. Il n'existe actuellement aucun traitement spécifique pour la maladie de Farber. Des corticoïdes peuvent être prescrits pour soulager la douleur. La greffe de moelle osseuse peut réduire les granulomes (petites masses de tissu enflammé) chez les personnes présentant une inflammation légère ou chez les patients sans complications pulmonaires ou du système nerveux. Chez les personnes âgées, les granulomes peuvent être rétrécis ou enlevés chirurgicalement.
Gangliosidose se compose de deux groupes différents de maladies génétiques. Les deux sont autosomiques récessifs et affectent également les hommes et les femmes.
Gangliosidose à GM1.
La gangliosidose à GM1 est causée par un déficit de l'enzyme bêta-galactosidase, entraînant une accumulation anormale de matières lipidiques acides, en particulier dans les cellules nerveuses du système nerveux central et périphérique systèmes. La gangliosidose à GM1 a trois manifestations cliniques :
- GM1 (le sous-type le plus grave, survenant peu après la naissance) peut inclure une neurodégénérescence, des convulsions, une hypertrophie du foie et de la rate, grossièreté des traits du visage, irrégularités du squelette, raideur articulaire, ballonnements, faiblesse musculaire, réaction de sursaut exagérée et problèmes de démarche. Environ la moitié des personnes touchées développent des taches rouge cerise dans les yeux. Les enfants peuvent être sourds et aveugles dès l'âge de 1 an et meurent souvent avant l'âge de 3 ans de complications cardiaques ou pneumonie.
- Enfant en retard La gangliosidose à GM1 débute généralement entre 1 et 3 ans. Les symptômes neurologiques comprennent l'ataxie, les convulsions, la démence et la difficulté à parler.
- Gangliosidose GM1 se développe entre 3 et 30 ans. Les symptômes comprennent une diminution de la masse musculaire (atrophie musculaire), des complications neurologiques moins graves et évoluant plus lentement que les autres formes troubles, opacité cornéenne chez certaines personnes et contractions musculaires soutenues qui provoquent des mouvements de torsion et répétitifs ou des postures anormales (dystonie). Les angiokératomes peuvent se développer dans la partie inférieure du tronc du corps. La taille du foie et de la rate est normale chez la plupart des personnes atteintes.
Gangliosidose à GM2.
La gangliosidose à GM2 amène également le corps à accumuler un excès de substances grasses acides dans les tissus et les cellules, principalement les cellules nerveuses. Ces troubles résultent d'un déficit de l'enzyme bêta-hexosaminidase. Les troubles GM2 comprennent :
- La maladie de Tay-Sachs (également connue sous le nom de variante B de la gangliosidose GM2) et ses variantes sont causées par une déficience de l'enzyme hexosaminidase A. Les enfants atteints se développent normalement au cours des premiers mois de la vie. Les symptômes commencent à l'âge de 6 mois et comprennent une perte progressive de la capacité mentale, une démence, une diminution du contact visuel, une augmentation une réaction de sursaut au bruit, une perte auditive progressive menant à la surdité, des difficultés à avaler, la cécité, des taches rouge cerise sur la rétine, et certains paralysie. Les crises peuvent commencer au cours de la deuxième année de la vie d'un enfant. Les bébés peuvent éventuellement devenir dépendants de la sonde d'alimentation et meurent souvent avant l'âge de 4 ans d'infections récurrentes. Malheureusement, il n'y a pas de traitement spécial. Les anticonvulsivants peuvent initialement contrôler les crises. D'autres traitements de soutien incluent une nutrition et une hydratation appropriées, ainsi que des méthodes pour garder les voies respiratoires ouvertes. Une forme rare de la maladie appelée maladie de Tay-Sachs à début tardif, survient chez les personnes âgées de 20 à 30 ans et se caractérise par une démarche instable et une détérioration neurologique progressive.
- La maladie de Sandhoff (Option AB) est une forme grave de la maladie de Tay-Sachs. Le début survient généralement à l'âge de 6 mois et n'est limité à aucun groupe ethnique. Les signes neurologiques peuvent inclure une détérioration progressive du système nerveux central, des faiblesse, cécité précoce, réaction de peur sévère au son, spasticité, choc ou contractions musculaires muscles (myoclonie), épilepsie, une tête anormalement agrandie (macrocéphalie) et des plaques rouges dans l'œil. D'autres symptômes peuvent inclure des infections respiratoires fréquentes, souffle au cœur, des traits du visage de poupée et une hypertrophie du foie et de la rate. Il n'y a pas de traitement spécifique pour la maladie de Sandhoff. Comme pour la maladie de Tay-Sachs, les soins de soutien comprennent le maintien des voies respiratoires ouvertes en tout temps, ainsi qu'une nutrition et une hydratation appropriées. Les anticonvulsivants peuvent initialement contrôler l'épilepsie (convulsions).
La maladie de Krabbe(aussi connu sous le nom leucodystrophie à cellules globulaires et lipidose galactosylcéramide) est une maladie autosomique récessive causée par un déficit de l'enzyme galactocérébrosidase. La maladie affecte le plus souvent les nourrissons avant l'âge de 6 mois, mais peut survenir à l'adolescence ou à l'âge adulte.
L'accumulation de graisses non digérées affecte la croissance de la gaine protectrice du nerf (gaine de myéline) et provoque une grave altération des capacités mentales et motrices. Les autres symptômes comprennent une faiblesse musculaire, une diminution de la capacité des muscles à s'étirer (hypotonie), une tension musculaire (spasticité), un choc soudain ou contractions musculaires des membres (crises myocloniques), irritabilité, fièvre inexpliquée, surdité, cécité, paralysie et difficulté à avaler. Une perte de poids à long terme peut également se produire.
La maladie peut être diagnostiquée par des tests enzymatiques et l'identification de son groupement cellulaire caractéristique en corps globoïdes dans la substance blanche du cerveau, démyélinisation des nerfs et dégénérescence, et destruction des cellules cérébrales. Chez les nourrissons, la maladie est généralement mortelle avant l'âge de 2 ans. Les personnes atteintes d'une forme plus avancée de la maladie ont une évolution plus douce de la maladie et vivent beaucoup plus longtemps.
Aucun traitement spécifique pour la maladie de Krabbe n'a été développé, bien qu'une greffe précoce de moelle osseuse puisse aider certaines personnes. Les personnes atteintes d'une forme plus avancée de la maladie ont une évolution plus douce de la maladie et vivent beaucoup plus longtemps.
Leucodystrophie métachromatique, ou MLD, est un groupe de troubles marqués par l'accumulation de substances grasses dans la substance blanche du système nerveux central et dans les nerfs périphériques et dans une certaine mesure dans les reins. Semblable à la maladie de Krabbe, la MLD affecte la myéline, qui recouvre et protège les nerfs. Cette maladie autosomique récessive est causée par une déficience de l'enzyme arylsulfatase A. Ce trouble touche aussi bien les hommes que les femmes.
La leucodystrophie métachromatique a trois formes caractéristiques: le nourrisson tardif, le juvénile et l'adulte.
- MLD infantile tardive commence généralement entre 12 et 20 mois après la naissance. Les enfants peuvent sembler normaux au début, mais ont des difficultés à marcher et une tendance à tomber, accompagnés de douleurs intermittentes dans les bras et les jambes. perte progressive de la vision conduisant à la cécité, des retards de développement et la perte des jalons précédemment acquis, des troubles de la déglutition, des convulsions et une démence jusqu'à 2 années. Les enfants développent également une fonte et une faiblesse musculaires progressives et finissent par perdre leur capacité à marcher. La plupart des enfants atteints de cette forme de maladie meurent avant l'âge de 5 ans.
- MLD juvénile commence généralement entre l'âge de 3 et 10 ans. Les symptômes comprennent une altération des performances scolaires, une déficience mentale, une ataxie, des convulsions et une démence. Les symptômes progressent, le décès survenant 10 à 20 ans après le début de la maladie.
- Forme adulte. Symptômes adultes début après l'âge de 16 ans et peut inclure ataxie, convulsions, tremblements anormaux des membres (tremblements), troubles de la concentration, dépression, troubles mentaux et démence. La mort survient généralement 6 à 14 ans après le début des symptômes.
La MLD ne peut pas être guérie. Le traitement est symptomatique et de soutien. La greffe de moelle osseuse peut dans certains cas retarder la progression de la maladie. Des progrès significatifs ont été réalisés avec la thérapie génique dans des modèles animaux atteints de MLD et dans les essais cliniques.
La maladie de Wolmanaussi connu sous le nom déficit en lipase acide lysosomaleest un trouble grave du stockage des lipides qui est généralement mortel à l'âge de 1 an. Cette maladie autosomique récessive est caractérisée par une accumulation d'esters de cholestérol (généralement la forme de transport du cholestérol) et triglycérides (la forme chimique sous laquelle les graisses existent dans le corps), qui peuvent s'accumuler de manière significative et endommager les cellules et tissus.
Les hommes et les femmes souffrent de ce trouble. Les nourrissons sont normaux et actifs à la naissance, mais développent rapidement une déficience mentale progressive, une hypertrophie du foie et une rate fortement hypertrophiée, des ballonnements et des problèmes gastro-intestinaux. jaunisse, anémie, vomissements et dépôts de calcium dans glandes surrénalesles faisant durcir.
Un autre type Le déficit en lipase acide lysosomale est maladie de stockage des esters de cholestérol. Cette maladie extrêmement rare survient à la suite du stockage d'esters de cholestérol et triglycérides dans les cellules sanguines, lymphe et tissu lymphoïde. Chez les enfants à l'âge adulte, le foie grossit, ce qui entraîne une cirrhose et chronique insuffisance hépatique. Les enfants peuvent également avoir des dépôts de calcium dans les glandes surrénales, et la jaunisse peut se développer dans les derniers stades de la maladie.
Actuellement, la question du remplacement enzymatique est activement étudiée à la fois dans la maladie de Wolman et dans l'accumulation d'un ester de cholestérol.
Attention! Les informations sont à titre informatif uniquement et ne sont pas destinées à diagnostiquer et à prescrire un traitement. Consultez toujours votre médecin spécialiste !