Hemophagocytic lymphohistiocytosis: mi ez, tünetek, kezelés, prognózis
Tartalom
- Mi a hemophagocyticus lymphohistiocytosis?
- jelek és tünetek
- Okok és kockázati tényezők
- Érintett populációk
- Tüneti rendellenességek
- Diagnosztika
- Standard kezelések
- Előrejelzés
Mi a hemophagocyticus lymphohistiocytosis?
Hemophagocyticus lymphohistiocytosis (HLH) ritka, életveszélyes állapot, amelyet hiperaktív, kóros immunválasz okoz. Az immunrendszer a szervezet természetes védekezőrendszere idegen vagy behatoló szervezetekkel vagy anyagokkal szemben. Az immunrendszer a sejtek, szövetek, szervek és fehérjék összetett hálózata, amelyek együttesen működnek a test egészségének megőrzése érdekében. A HLH -ban az immunrendszer reagál egy ingerre vagy kiváltóra, gyakran fertőzésre, de a válasz hatástalan és kóros. Ez a hatástalan, rendellenes reakció számos jelet és tünetet okoz, amelyek kezelés nélkül életveszélyessé válhatnak. Egyes betegek genetikai hajlamúak lehetnek a hemophagocyticus lymphohistiocytosis kialakulására. Ezt elsődleges vagy családi formának nevezik. Másoknál a betegség szórványosan fordul elő, általában hajlamos betegséggel vagy rendellenességgel. Másodlagos formának nevezik. A másodlagos formák gyakoribbak, mint a családi formák. A hemophagocyticus lymphohistiocytosis leggyakrabban a csecsemőket érinti születéstől 18 hónapos korig, de bármilyen korú embert érinthet. A korai diagnózis és azonnali kezelés elengedhetetlen.
A hemophagocytic lymphohistiocytosis (HLH) olyan állapot, amelynek különböző okai vannak. Ennek az állapotnak a leírására több nevet is használnak. A családi hemophagocyticus lymphohistiocytosis (familiáris HLH) olyan genetikai formákra utal, amelyeket kóros génváltozat okoz. 2018 áprilisától több gén rendellenességet azonosítottak okként. A makrofágaktivációs szindróma (CAM) egy olyan kifejezés, amelyet az autoimmun vagy autoinflammatorikus betegségben szenvedő embereknél előforduló hemofagocitás lymphohistiocytosis leírására használnak. Ez egyfajta másodlagos HLH. A CAM -hoz leggyakrabban társuló betegségek fiatalkori szisztémásak ízületi gyulladás, felnőtt Still -betegség és szisztémás lupus erythematosus.
jelek és tünetek
A hemophagocyticus lymphohistiocytosis kialakulása és súlyossága személyenként nagyon eltérő lehet. A kialakuló specifikus tünetek is nagymértékben változhatnak, bár a betegség gyakran több szerv károsodását okozza. Általában a betegeknél láz, kiütés, májnagyobbodás jelentkezik (hepatomegalia) és növekszik lép (splenomegalia). A láz elhúzódó és tartós is lehet, gyakran az antibiotikumokra adott válasz nélkül. Néha a nyirokcsomók is megnagyobbodnak (limfadenopátia). A nyirokcsomók a nyirokrendszer, az erek, a csatornák és a csomópontok részei, amelyek szűrnek és elosztanak bizonyos fehérjében gazdag (nyirok) és vérsejteket test. A nyirokcsomók kisméretű szerkezetek, amelyek az egész testben klaszterekben helyezkednek el, és segítenek a káros anyagok szűrésében vagy eltávolításában a szervezetből.
Ezeket a kezdeti jeleket és tüneteket nem specifikusnak írják le. Ez azt jelenti, hogy ezek a jelek és tünetek sok más betegségre vagy állapotra jellemzőek, ami megnehezítheti a helyes diagnózis felállítását.
A betegeknél alacsony lehet a keringő vörösvértestek szintje is (anémia) és a keringő vérlemezkék alacsony szintje (thrombocytopenia). A vörösvértestek oxigént szállítanak a szervezetbe, és a vérlemezkék lehetővé teszik a szervezet számára, hogy vérrögöket (vérrögöket) képezzenek, amelyek megállítják a vérzést. A vérszegénységben szenvedő betegek fáradtságot, fokozott alvási szükségletet, gyengeséget, szédülést, ingerlékenységet, fejfájást, sápadt bőrt, légszomjat tapasztalhatnak (légszomj) és szívproblémák. A thrombocytopeniában szenvedő betegek hajlamosabbak a minimális trauma utáni véraláfutásokra és a nyálkahártyák, különösen az íny és az orr spontán vérzésére.
Néhány embernél neurológiai tünetek alakulhatnak ki, beleértve epilepszia, a mentális állapot és az ingerlékenység megváltozása, bizonyos koponyaidegek bénulása és az önkéntes mozgások koordinációjával kapcsolatos problémák (ataxia). A betegeknél fennáll a veszélye a posterior reverzibilis encephalopathia szindróma kialakulásának, amely gyors fejfájást, megváltozott tudatosságot, görcsöket és látásromlást okoz. Neurológiai problémák leggyakrabban családi hemophagocyticus lymphohistiocytosis esetén jelentkeznek.
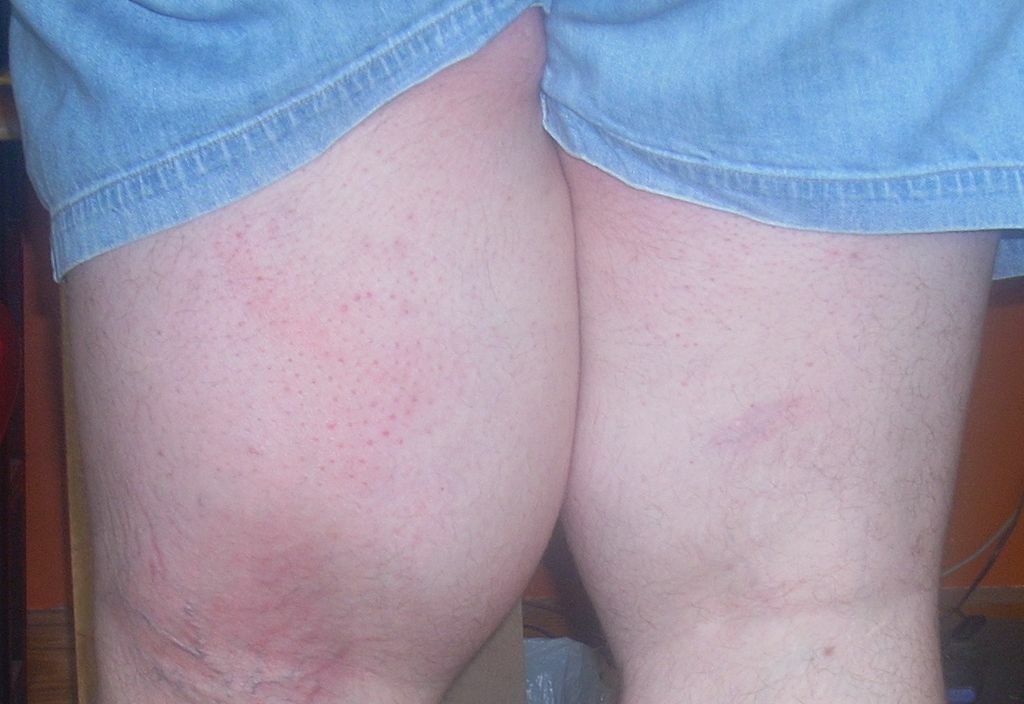
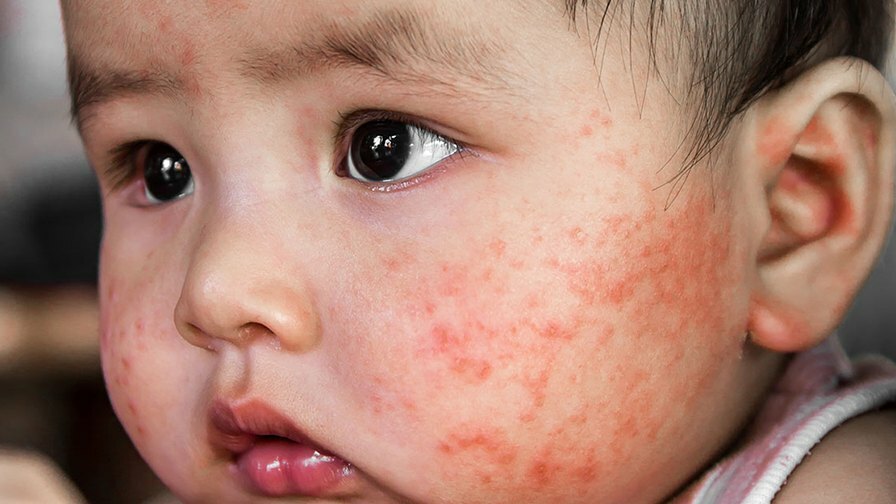
További tünetek jelentkezhetnek a személy sajátos szervrendszerétől függően. Ezek a tünetek jelentős légzési problémákat (tüdőfunkciózavar), súlyos alacsony szintet tartalmazhatnak vérnyomás (hipotenzió), májgyulladás (hepatitis), veseműködési zavar, a bőr és a fehérjék besárgulása szem (sárgaság), ödéma folyadék felhalmozódása miatt, puffadás folyadék felhalmozódása miatt (hasi ascites) és különböző bőrproblémák, beleértve a bőr gyulladás miatti széles körű vörösödését (eritroderma), kiütéseket, vérfoltokat (purpura) és apró foltokat a bőrön (petechiák).
Olvassa el még:Fenilketonuria
Okok és kockázati tényezők
A hemophagocyticus lymphohistiocytosis elsődleges és másodlagos (szerzett) formákra oszlik. Az állapot az immunrendszer hatástalan, rendellenes válaszából ered egy ingerre vagy kiváltóra. A jelek és tünetek kialakulásának mögöttes mechanizmusai összetettek. Van túltermelés és hiperaktív immunrendszeri sejtek, az úgynevezett hisztiociták és T -sejtek. Ezek a fehérvérsejtek típusai, amelyek az immunrendszer elsődleges sejtjei, és segítik a szervezetet a fertőzés elleni küzdelemben.
A hisztiociták (más néven makrofágok) nagy fagocita sejtek, amelyek általában szerepet játszanak a fertőzésre és sérülésre adott válaszban. A fagocita sejt minden olyan sejt, amely elnyeli és elpusztítja a behatoló mikroorganizmusokat vagy sejtmaradványokat. A makrofágok citokineket is termelnek, olyan fehérjéket, amelyek stimulálják vagy elnyomják az immunrendszer más sejtjeit, és elősegítik a gyulladást a betegségre adott válaszként. A citokinek túltermelése végül súlyos szövetkárosodáshoz vezet. A makrofágok tévesen elnyelhetik és elpusztíthatják az egészséges szöveteket, beleértve az egészséges vérsejteket is, hemofagocitózisnak. A citotoxikus limfociták, beleértve a T -sejteket és a természetes gyilkos sejteket, nem működnek megfelelően. Ezek a sejtek megszüntetnek más sérült, stresszes vagy fertőzött sejteket. A HLH -ban a citotoxikus limfociták nem tudják megölni az aktivált makrofágokat, ami lehetővé teszi számukra kórosan halmozódnak fel a szervezet szerveiben és szöveteiben, ami tovább aktiválja ezt a hatástalanságot immunválasz. Ezek az immunrendszeri rendellenességek okozzák a túlzott gyulladást és a szövetek pusztulását, amelyek ezt az állapotot jellemzik.
- Elsődleges hemophagocyticus lymphohistiocytosis.
A HLH elsődleges formája bizonyos gének kóros variánsaihoz kapcsolódik. A gének utasításokat adnak olyan fehérjék előállítására, amelyek kritikus szerepet játszanak számos testi funkcióban. Amikor génmutáció következik be, a fehérjetermék hibásan működhet, nem hatékony, hiányzik vagy túltermelődik. Egy adott fehérje funkcióitól függően befolyásolhatja a szervezet számos szervrendszerét.
Legalább négy különböző gént azonosítottak, amelyek genetikai hajlamhoz vezetnek a HLH kialakulására. A genetikai hajlam azt jelenti, hogy egy személy rendelkezik egy bizonyos rendellenesség génjével vagy génjeivel, de a rendellenesség csak akkor alakul ki, ha más tényezők segítik a rendellenesség kiváltását. Négy gént azonosítottak - ezek PRF1 (családi hemofagocitás limfocitózis 2 -es típus), UNC13D (családi 3. típusú hemofagocitás limfocitózis), STX11 (4 -es típusú családi hemofagocitás limfocitózis és STXBP2 (családi hemofagocitás limfocitózis 1 -es típus).
Ezek a gének olyan fehérjéket termelnek, amelyek fontos szerepet játszanak az immunrendszerben. Szerepük van az aktivált immunsejtek leállításában vagy elpusztításában, amikor már nincs rájuk szükség. Ezeknek a géneknek a variációi (mutációi) miatt a gének nem termelnek elég vagy nem hatékony változatokat ezekből a fehérjékből. Ennek eredményeképpen az aktivált immunsejtek, amelyeket általában le kell tiltani vagy el kell pusztítani, továbbra is működnek, és továbbra is működnek, végül károsítva az egészséges sejteket és szöveteket.
A genetikai betegségeket az apától és az anyától kapott kromoszómákon található, egy adott tulajdonsághoz tartozó gének kombinációja határozza meg. A recesszív mintázatú öröklött rendellenességek akkor fordulnak elő, amikor egy személy ugyanazt a tulajdonságot ugyanazt a variáns gént örökölte minden szülőtől. Ha egy személy egy normális gént és egy gént kap a betegséghez, akkor a betegség hordozója lesz, de általában tünetmentes. A hordozó szülők két kockázata, hogy a hibás gént továbbítják, és így beteg gyermeket szülnek, minden terhesség esetén 25%. Minden terhesség esetén 50% a kockázata annak, hogy gyermeke lesz, aki hordozó lesz, mint a szülők. 25%annak a valószínűsége, hogy a gyermek mindkét szülőtől normális géneket kap, és genetikailag normális lesz. A kockázat férfiaknál és nőknél azonos.
Néhány embernek különböző változatai lehetnek, amelyek befolyásolják a betegség egyik génjének minden egyes példányát (komplex heterozigóták), míg másoknak digenikus öröklődésük lehet. A digenikus öröklődés azt jelenti, hogy kóros variánsuk van két különböző génben, amelyekről ismert, hogy hemofagocitás limfohistiocitózissal járnak együtt.
Olvassa el még:Waardenburg szindróma
Néhány betegnél a hemophagocyticus lymphohistiocytosis egy tágabb genetikai rendellenesség része. Ezek a rendellenességek közé tartozik a 2 -es típusú Griszelli -szindróma, Chédiak-Higashi szindróma, X-kapcsolt limfoproliferatív szindróma, interleukin-2-indukálta T-sejt-kináz hiánya, hiány CD27, germán-Pudlak szindróma, lizinurikus fehérje intolerancia és krónikus granulomatózus betegség. Néhány betegnél a hemophagocyticus lymphohistiocytosis lehet az egyetlen klinikai probléma.
- Másodlagos hemophagocyticus lymphohistiocytosis.
Másodlagos (vagy szerzett) HLH -ban szenvedő betegeknél a betegség az immunrendszer fokozott kóros reakciója miatt alakul ki, ami ismeretlen okok miatt következik be. Ennek a rendellenességnek nincs családi előzménye, és nem ismert genetikai tényezők azonosíthatók. A másodlagos hemophagocyticus lymphohistiocytosishoz vezető állapotok közé tartoznak a vírusfertőzések, különösen Epstein-Barr vírusegyéb fertőzések, beleértve a bakteriális, vírusos és gombás fertőzéseket, legyengült vagy elnyomott immunrendszert, autoimmun betegség, autoinflammatorikus betegségek, reumatológiai betegségek, például juvenilis idiopátiás ízületi gyulladás, anyagcserezavarok. és a rákhoz hasonló non-Hodgkin limfóma.
Az, hogy ezek a hajlamosító állapotok pontosan hogyan okoznak jeleket és tüneteket, és különösen azt, hogyan hatástalan, kóros immunválaszt okozhat hemofagocitás limfohistiocitózisban, nem teljesen tanult.
Érintett populációk
A hemophagocyticus lymphohistiocytosis leggyakrabban csecsemőket vagy kisgyermekeket érint, de bármilyen korú embereket érinthet. A betegség a fiúkat és a lányokat egyaránt érinti. A felnőtt férfiak valamivel valószínűbbek, mint a nők. A pontos gyakoriság és elterjedtség ismeretlen. A ritka rendellenességeket gyakran rosszul vagy rosszul diagnosztizálják, ami megnehezíti a valódi gyakoriság meghatározását az általános populációban. Az ilyen állapotú emberek körülbelül 25% -a családos.
Tüneti rendellenességek
A következő betegségek tünetei hasonlóak lehetnek a hemophagocyticus lymphohistiocytosis tüneteihez, és nagyon fontos megkülönböztetni ezeket a rendellenességeket tőle.
Sok különböző állapotnak lehetnek olyan jelei és tünetei, amelyek hasonlóak a hemophagocyticus lymphohistiocytosishoz. Ezek tartalmazzák
- Di Georgi szindróma (Di Georgie);
- Kawasaki betegség;
- tuberkulózis;
- leishmaniasis;
- többszörös szervi diszfunkció szindróma;
- agyvelőgyulladás;
- gyógyszerreakció eozinofíliával és szisztémás tünetekkel (DRESS -szindróma);
- autoimmun limfoproliferatív szindróma;
- thromboticus thrombocytopenia purpura;
- hemolitikus urémiás szindróma.
Májbetegség és egyes fertőzések hasonlíthatnak a hemophagocyticus lymphohistiocytosisra is.
Diagnosztika
A diagnózis a jellegzetes tünetek azonosításán, a beteg részletes kórtörténetén, az alapos klinikai értékelésen és a különféle speciális teszteken alapul. Iránymutatásokat tettek közzé, amelyek részletezik a HLH diagnosztizálásához szükséges kritériumokat. Ha a következő nyolc tünet közül öt jelen van, klinikai diagnózist lehet felállítani.
Ez a nyolc tünet a következő:
- láz;
- a lép megnagyobbodása (splenomegalia);
- alacsony vörösvértestek, fehérvérsejtek vagy vérlemezkék (cytopenia);
- kórosan magas trigliceridnek nevezett zsír szintje a vérében (hipertrigliceridémia) vagy bizonyos véralvadási fehérje alacsony szintje (hipofibrinogenémia);
- a vérsejtek pusztulása a makrofágok által (hemofagocitózis) a csontvelőben;
- a természetes gyilkos sejtek alacsony vagy hiányzó aktivitása;
- A vashoz kötődő fehérje kórosan magas vérszintje (ferritinémia)
- az oldható interleukin-2 receptor (sCD25) emelkedett szintje, egy speciális fehérje, amely az immunrendszer stimulálása során felhalmozódik a vérben.
Mivel a hemophagocyticus lymphohistiocytosis tünetei nem specifikusak, a betegek gyakran túlélhetik a hosszú távú betegségeket, és kórházba kerülhetnek a diagnózis előtt.
- Elemzések.
Az orvosok vérvizsgálatokat rendelhetnek el a teljes vérsejtszám meghatározásához, amelyek a vörösvérsejtek, a fehérvérsejtek és a vérlemezkék szintjét mérik. A vérvizsgálatok abnormálisan magas ferritin- vagy magas trigliceridszintet is kimutathatnak. Az orvosok vérvizsgálatokkal is megkereshetik a vérben a fertőzés jeleit, és teszteket végezhetnek annak megállapítására, hogy mennyire jól alakulnak ki a vérrögök (alvadási vizsgálatok). Az orvosok olyan vizsgálatokat is rendelhetnek, amelyek felmérhetik a máj egészségét és működését.
Néha egy csontvelő biopszia (sebészeti eltávolítás és szövetminta mikroszkópos vizsgálata) és vizsgálja meg a hemofagocitózis jeleit, a fertőzés vagy a fertőző szervezetek jeleit vagy a torlódást makrofágok.
A molekuláris genetikai vizsgálatok egyes betegeknél megerősíthetik a hemophagocyticus lymphohistiocytosis diagnózisát. A molekuláris genetikai vizsgálatok mutációkat tudnak kimutatni az ismert négy specifikus gén egyikében ennek a rendellenességnek a családi formáit okozzák, de csak szakosodott diagnosztikai szolgáltatásként érhető el laboratóriumok.
Olvassa el még:Pancytopenia
Standard kezelések
A hemophagocyticus lymphohistiocytosis kezelése az egyes személyekben megnyilvánuló specifikus tünetek kiküszöbölésére irányul. A kezeléshez szükség lehet egy szakembercsoport összehangolt erőfeszítéseire. Gyermekorvosok, vérbetegségek diagnosztizálásával és kezelésével foglalkozó szakemberek (hematológusok), rák diagnosztizálásával és kezelésével foglalkozó szakemberek (onkológusok), betegségek diagnosztizálásával és kezelésével foglalkozó szakemberek immunrendszer (immunológusok), genetikusok (családi formák esetén), szociális munkások és más egészségügyi szakemberek szisztematikus és integrált tervezést igényelhetnek kezelés. A pszichoszociális támogatás is fontos az egész család számára. A genetikai tanácsadás hasznos lehet az érintettek és családjaik számára.
A specifikus terápiás eljárások és beavatkozások sok tényezőtől függően változhatnak, például a kiváltó októl; bizonyos tünetek jelenléte vagy hiánya; a tünetek és rendellenességek általános súlyossága; egy személy kora és általános egészségi állapota; és / vagy más elemek. Döntéseket kell hozni az egyes gyógyszeres kezelési rendek és / vagy egyéb kezelések alkalmazásával kapcsolatban az orvosok és az orvosi csapat más tagjai, miután a beteggel alaposan konzultáltak, sajátosságai alapján ügy; a lehetséges előnyök és kockázatok alapos megvitatása, beleértve a lehetséges mellék- és hosszú távú hatásokat; a beteg preferenciái; és egyéb releváns tényezők.
Azok az áldozatok, akiknek általános egészségi állapota kellően erős, kezelhetők az alapbetegség miatt, például egy mögöttes fertőzés kezelésére szolgáló gyógyszerek vagy az autoimmun betegségek megfelelő kezelése, vagy rák. Az alapbetegség kezelése eltávolíthatja azt a kiváltó okot, amely az immunrendszer rendellenes válaszához vezetett.
Azok az áldozatok, akiknek egészségi állapota romlik, azonnali, a HLH -ra specifikus kezelésre szorulnak. 1994-ben a Histiocyte Society iránymutatásokat tett közzé ennek az állapotnak a kezelésére (HLA-94). 2004-ből (HLA-2004) is publikáltak tanulmányokat, amelyek kissé eltértek.
Ezek a kezelési rendek magukban foglalják a kemoterápiát és az immunrendszert elnyomó gyógyszereket (immunszuppresszánsok). Az immunrendszer túlműködő sejtjeit célozzák meg és pusztítják el, csökkentve a hemofagocitás lymphohistiocytosisra jellemző életveszélyes gyulladást.
A kezdeti kezelés után, amely körülbelül 8 hétig tart, a betegeket fokozatosan lekapcsolják más gyógyszerekről. Ha az áldozatok rosszul reagáltak erre a kezelésre, javasolható allogén őssejt -transzplantáció. Ez a kezelés azoknak is ajánlott, akiknél ismert HLH gének mutációi vannak, központi idegrendszeri rendszer és vérrák (hematológiai rosszindulatú daganat), amely nem reagál kezelés.
Az allogén őssejt -transzplantáció olyan eljárás, amelyben az érintett személy őssejtjeit megfelelő egészséges donor őssejtjeire cserélik. Az őssejtek a csontvelő speciális sejtjei, amelyek különböző típusú vérsejteket termelnek (például vörösvértesteket, fehérvérsejteket, vérlemezkéket).
A betegek nagy dózisú kemoterápiát vagy sugárzást kapnak az őssejtek elpusztítására. Az őssejteket ezután donor sejtekkel helyettesítik. Az allogén őssejt-transzplantáció magas kockázatú eljárás, lehetséges mellékhatásokkal.
Néhány embernek szüksége lehet vérátömlesztésre, mert alacsony a keringő vörösvértestek vagy vérlemezkék szintje. Egyes orvosok antibiotikumokat javasolhatnak a fertőzés megelőzésére (profilaktikus kezelés).
2018 -ban engedélyezték a Gamifant (emapalumab) elsődleges HLH -ban szenvedő gyermek- és felnőtt betegek kezelésére akik tűzálló, visszatérő vagy progresszív betegségben szenvednek, vagy akik nem tolerálják a hagyományosat HLH terápia.
Előrejelzés
A teljes halálozási arány 50%. A kedvezőtlen prognosztikai tényezők közé tartozott a rosszindulatú daganatokkal összefüggő HLH a betegek fele 1,4 hónap után halt meg, szemben a 22,8 hónappal azoknál a betegeknél, akiknél a HLH nem társult tumor.
Egyes esetekben a másodlagos HLH önkorlátozható lehet, mivel a betegek teljesen gyógyuljon, miután csak támogató ellátást kapott (pl. csak intravénásan) immunglobulin). A citotoxikus és immunszuppresszív terápia alkalmazása nélküli hosszú távú remisszió azonban nem valószínű a legtöbb HLH -ban szenvedő felnőttnél és a központi idegrendszeri betegeknél (fej és / vagy gerinc) agy).