
מחלות אחסון ליזוזומליות: מה זה, תסמינים, טיפול, פרוגנוזה
תוֹכֶן
- מהן מחלות אחסון ליזוזומליות?
- מהם שומנים?
- כיצד עוברות מחלות אחסון ליזוזומליות בתורשה?
- סוגים ותסמינים של מחלות אחסון ליזוזומליות
מהן מחלות אחסון ליזוזומליות?
מחלות אחסון ליזוזומליות, הם קבוצה של מחלות מטבוליות תורשתיות בהן מצטברות כמויות מזיקות של חומרי שומן (שומנים) בתאים וברקמות שונות של הגוף.
אנשים הסובלים מהפרעות אלו או שאינם מפיקים מספיק מאחד האנזימים הדרושים לפירוק (מטבוליזם) של שומנים, או שהם מייצרים אנזימים שאינם פועלים כראוי.
עם הזמן, הצטברות מוגזמת זו של שומן עלולה לגרום לנזק תמידי לתאים ורקמות, במיוחד במוח, במערכת העצבים ההיקפית (עצבים מחוט השדרה לשאר חלקי גוּף), כָּבֵד, טְחוֹל ומח עצם.
מהם שומנים?

שומנים הם חומרים דמויי שומן המהווים חלקים חשובים של הממברנות בתוך ובין התאים ובין מעטפת המיאלין, המכסה ומגן על העצבים. השומנים כוללים שמנים, חומצות שומן, שעוות, סטרואידים (כגון כולסטרול ואסטרוגן), ותרכובות נלוות אחרות.
חומרים שומניים אלה מאוחסנים באופן טבעי בתאים, באיברים וברקמות הגוף. גופים זעירים בתוך תאים הנקראים ליזוזומים ממירים או מטבוליזים באופן קבוע שומנים וחלבונים לרכיבים קטנים יותר כדי לספק אנרגיה לגוף. הפרעות בהן חומר תוך תאי, שאינו ניתן לעיכול, נשאר בליזוזומים, נקראות מחלות אחסון ליזוזומליות.
בנוסף למחלות הקשורות להצטברות שומנים, מחלות ליזוזומליות אחרות הקשורות להצטברות כוללות מוקוליפידוזות, בהן מצטברים תאים ורקמות. כמות מוגזמת של שומנים עם מולקולות סוכר המחוברות אליהם, כמו גם מוקופוליסכרידוזים, שבהם מצטברת כמות מוגזמת של סוכר גדול ומורכב. מולקולות.
כיצד עוברות מחלות אחסון ליזוזומליות בתורשה?

מחלות אחסון ליזוזומליות עוברות בתורשה מאחד ההורים או שניהם הנושאים גן פגום המווסת אנזים ספציפי של חילוף חומרים בשומנים במחלקה של תאים בגוף. הם יכולים לעבור בירושה בשתי דרכים:
- ירושה אוטוזומלית רצסיבית מתרחשת כאשר שני ההורים נושאים ומעבירים העתק של הגן הפגום, אך אף אחד מההורים אינו מושפע מההפרעה. לכל ילד שנולד להורים אלה יש 25 אחוז סיכוי לרשת את שני העותקים של הגן הפגום, 50 אחוז סיכוי להיות נשא דמוי הורים וסיכוי של 25 אחוזים לא לרשת כל עותק של הפגום גֵן. ילדים משני המינים יכולים לעבור בירושה באופן אוטוסומלי רצסיבי.
- תורשה רצסיבית צמודה ל- X (או הקשורה למין) מתרחשת כאשר האם נושאת את הגן המושפע על כרומוזום X. כרומוזומי X ו- Y מעורבים בקביעת מין. לנשים יש שני כרומוזומי X, בעוד שלגברים יש כרומוזום X אחד וכרומוזום Y אחד. לבני נשאים יש 50 אחוז סיכוי לרשת את ההפרעה ולהשפיע עליהם, שכן בנים מקבלים כרומוזום X אחד מאמם וכרומוזום Y אחד מאביהם. לבנות יש סיכוי של 50 אחוז לרשת כרומוזום X מושפע מאמם והן נשאיות או מושפעות קלות. גברים מושפעים אינם מעבירים את המחלה לבניהם, אך בנותיהם יהיו נשאות ונשאות של המחלה.
סוגים ותסמינים של מחלות אחסון ליזוזומליות
מחלת גושה - השכיחה ביותר ממחלות האחסון הליזוזומליות. המחלה נגרמת על ידי מחסור באנזים glucocerebrosidase, מה שמוביל להצטברות של glucocerebroside ברקמות רבות. חומר שומני יכול להצטבר במוח, הטחול, הכבד, הכליות, הריאות ומח העצם.
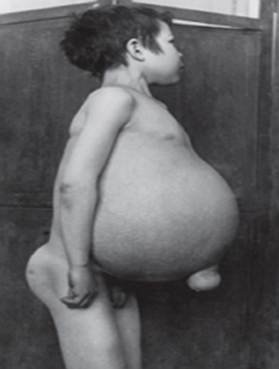
התסמינים עשויים לכלול נזק מוחי, הגדלת הטחול וכבד, תפקוד כבד לקוי, הפרעות בשלד ופגיעה בעצמות העלולות לגרום לכאבים ושברים, נפיחות של בלוטות הלימפה ומפרקים סמוכים (לפעמים), נפיחות, גוון עור חום, אֲנֶמִיָה, טסיות דם וכתמים צהובים על העיניים.
האנשים שנפגעו בצורה החמורה ביותר עלולים גם הם להיות רגישים יותר לזיהומים. המחלה פוגעת בגברים ובנשים באופן שווה.
למחלת גושה שלוש תת סוגים קליניים כלליים:
- סוג 1 (או לא נוירונופתי סוג) היא הצורה הנפוצה ביותר של המחלה בארצות הברית ובאירופה. המוח אינו מושפע, אך עלולה להיות פגיעה בריאות ובמקרים נדירים בכליות. התסמינים יכולים להתחיל בשלב מוקדם של החיים או בבגרותם ולכלול כבד מוגדל והגדלה חמורה של הטחול, מה שעלול להוביל לקרע ולסיבוכים נוספים. חולשת השלד ומחלות עצמות יכולות להיות נרחבות. אנשים בקבוצה זו בדרך כלל חוברים בקלות בשל מספר הטסיות הנמוך בדם. הם עשויים לחוות גם עייפות עקב אנמיה. בהתאם להופעת המחלה וחומרתה, אנשים עם סוג 1 יכול לחיות עד הבגרות. חולים רבים סובלים ממחלה קלה או שאינם מראים סימפטומים כלשהם. למרות ש 1סוּג מחלת גושה נפוצה בקרב בני המורשת היהודית האשכנזית ויכולה להשפיע על אנשים מכל מוצא אתני.
- סוג 2 (אוֹ נוירופתית חריפה בילדות מחלת גושה) מתחילה בדרך כלל תוך 3 חודשים מיום הלידה. התסמינים כוללים נזק מוחי מתקדם ומתקדם, ספסטיות, התקפים, גפיים נוקשות, כבד מוגדל (hepatomegaly הכבד) וטחול (splenomegaly), תנועות עיניים חריגות ויכולת יניקה ובליעה ירודה. ילדים מושפעים מתים בדרך כלל לפני גיל שנתיים.
- סוג 3 (נוירונופתי כרוני צורה) יכול להתחיל בכל עת במהלך הילדות או אפילו בבגרות. הוא מאופיין בסימפטומים נוירולוגיים מתקדמים לאט אך פחות חמורים בהשוואה למחלת גושה חריפה 2 סוגים. התסמינים העיקריים כוללים הפרעות בתנועת העין, ליקויים קוגניטיביים, תיאום לקוי, התקפים, הגדלת הטחול ו / או הכבד, הפרעות בשלד, הפרעות דם כולל אנמיה ובעיות עם נְשִׁימָה. כמעט כולם עם מחלת גושה 3 סוגים, שמקבל טיפול החלפת אנזים יגיע לבגרות.
קראו גם:תסמיני חצבת אצל ילדים, צילום מהשלב הראשוני
למטופלים סוג ראשון ושלישי טיפול החלפת אנזים הניתן תוך ורידי כל שבועיים יכול להפחית באופן דרמטי את גודל הכבד והטחול, להפחית הפרעות בשלד ולהפוך ביטויים אחרים. השתלת מח עצם מוצלחת מטפלת בתופעות הנוירולוגיות של המחלה. עם זאת, הליך זה טומן בחובו סיכונים משמעותיים ולעתים נדירות הוא מתבצע אצל אנשים הסובלים ממחלת גושה.
במקרים נדירים, ייתכן שיהיה צורך בניתוח להסרת כל או חלקו טחול (אם יש לאדם ספירת טסיות נמוכה מאוד או כאשר איבר מוגדל משפיע מאוד על נוחות האדם). עירוי דם עשוי להועיל לחלק מהאנשים הסובלים מאנמיה. אחרים עשויים להזדקק לניתוח החלפת מפרקים כדי לשפר את הניידות ואיכות החיים. כיום אין טיפול יעיל בנזק המוח שיכול להתרחש אצל אנשים הסובלים ממחלת גושה מסוג 2 וסוג 3.
מחלת נימן פיק היא קבוצה של הפרעות רצסיביות אוטוזומליות הנגרמות על ידי הצטברות שומן וכולסטרול בתאי הכבד, הטחול, מח העצם, הריאות ובמקרים מסוימים המוח.
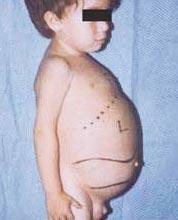
סיבוכים נוירולוגיים יכולים לכלול אטקסיה (חוסר תיאום שרירים שיכול להשפיע על הליכה, כתיבה ואכילה, בין פונקציות אחרות), שיתוק עיניים, ניוון מוח, בעיות למידה, ספסטיות, קשיי האכלה ובליעה, עיוות דיבור, אובדן טונוס שרירים, רגישות יתר למגע, וחוסר אטימות בקרנית עקב הצטברות מופרזת חומרים. אצל 50% מהאנשים שנפגעו, הילה אופיינית אדומה-דובדבן מתפתחת סביב מרכז הרשתית, אותה יכול לראות רופא עם מכשיר מיוחד.
מחלת נימן פיק מסווגת לשלושה תת סוגים כלליים:
- סוג א, הצורה החמורה ביותר, מתחילה בילדות המוקדמת. תינוקות נראים תקינים בלידה, אך עד שישה חודשים מתפתחת נזק מוחי עמוק, כבד וטחול מוגדלים ובלוטות לימפה ובלוטות מתחת לעור (קסנטומות). הטחול עשוי להתרחב פי 10 מהגודל הרגיל שלו ועלול לקרע ולגרום לדימום. ילדים אלה נעשים חלשים יותר ויותר, מאבדים תפקוד מוטורי, עלולים להפוך לאנמיים, והם מועדים לזיהומים חוזרים. לעתים רחוקות הם חיים יותר מ -18 חודשים. צורה זו של המחלה שכיחה ביותר במשפחות יהודיות.
- סוג ב(או הופעת צעירים) בדרך כלל אינו משפיע על המוח, אך רוב הילדים מפתחים אטקסיה, פגיעה בעצבים היוצאים מחוט השדרה (נוירופתיה היקפית), וקשיים ריאתיים המתקדמים עם הגיל. הגדלת הכבד והטחול אופיינית למתבגרים. אנשים עם סוג ב יכול לחיות יחסית זמן, אך רבים דורשים טיפול בחמצן לכל החיים עקב נזק לריאות. Niemann-Pick מסוג A ו- B הם תוצאה של הצטברות של חומר שומני בשם ספינגומיאלין עקב מחסור באנזים הנקרא ספינגומילינאז.
- סוג C עשוי להופיע מוקדם בחיים או להתפתח במהלך גיל ההתבגרות או אפילו בבגרות. מחלת נימן פיק סוג ג אינו נגרם על ידי מחסור בספלינגומילינאז, אלא מחוסר בחלבוני NPC1 או NPC2. כתוצאה מכך, שומנים שונים ובעיקר כולסטרול מצטברים בתאי עצב וגורמים לתפקודם. מעורבות המוח יכולה להיות נרחבת, וכתוצאה מכך חוסר יכולת להסתכל למעלה ולמטה, קושי בהליכה ובליעה, אובדן שמיעה מתקדם והתקדמות. דמנציה. אנשים עם סוג ג בעלי הגדלה קלה בלבד של הטחול והכבד. תוחלת חיים של אנשים עם סוג ג משתנה במידה ניכרת. חלק מהאנשים מתים בילדות, בעוד שאחרים שנפגעים פחות עשויים לחיות גם בבגרותם.
כיום אין תרופה למחלת נימן פיק. הטיפול תומך. ילדים בדרך כלל מתים מזיהום או מאובדן נוירולוגי מתקדם. היו מקרים של השתלת מח עצם במספר חולים עם סוג ב עם תוצאות מעורבות.
מחלת פאברי ידוע גם כ מחסור באלפא-גלקטוזידאז A, גורם להצטברות של חומר שומני במערכת העצבים האוטונומית (אותו חלק של מערכת העצבים ש שולט בתפקודים לא רצויים כגון נשימה ודופק), בעיניים, בכליות ובקרדיווסקולאר מערכת.
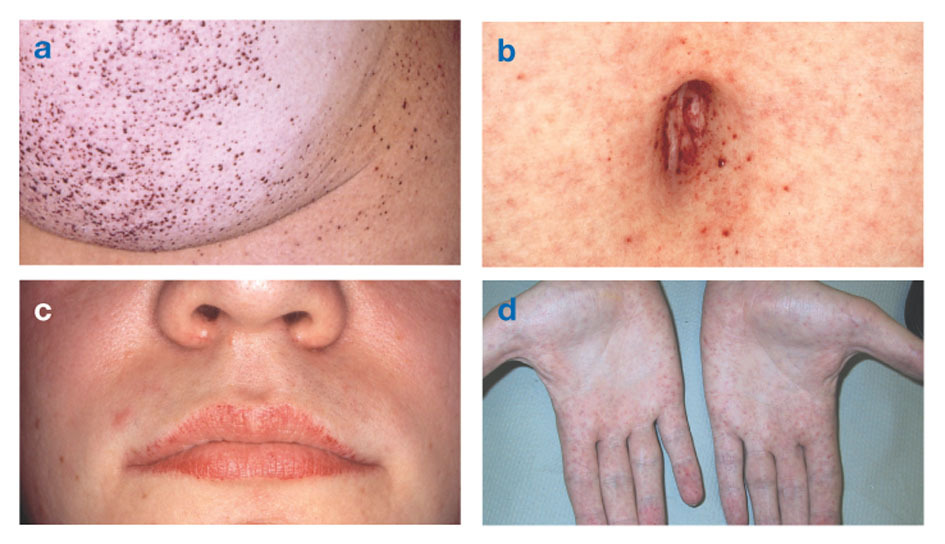
מחלת פאברי היא מחלת השומנים היחידה הקשורה ל- X. המחלה פוגעת בעיקר בגברים, אם כי אצל נשים מתרחשת צורה מתונה ומשתנה יותר. לעיתים נדירות, לנשים מושפעות יש סימפטומים חמורים הדומים לאלה שנראים אצל גברים הסובלים מההפרעה. הופעת התסמינים מתרחשת בדרך כלל במהלך הילדות או ההתבגרות.
סימנים נוירולוגיים כוללים כאבי צריבה בידיים וברגליים המחמירים במזג אוויר חם או לאחר מכן פעילות גופנית, כמו גם הצטברות של עודפי חומר בשכבות השקופות של הקרנית (מה שמוביל לעיכוב, אך ללא שינויים בראייה). הצטברות שומן בדפנות כלי הדם עלולה לשבש את זרימת הדם ולסכן אדם שבץ אוֹ התקף לב. תסמינים אחרים כוללים לב מוגדל, פרוגרסיבי כשל כלייתיהמוביל למחלת כליות סופנית, בעיות במערכת העיכול, ירידה בהזעה וחום.
קראו גם:רככת אצל תינוקות: סיבות, תסמינים וטיפול
אנגיוקראטומות (כתמים קטנים, לא סרטניים, אדומים-סגולים על העור) יכולים להתפתח בפלג הגוף התחתון ולהיות נפוצים יותר עם הגיל.
אנשים הסובלים ממחלת פאברי מתים לעיתים קרובות בטרם עת כתוצאה מסיבוכים אִי סְפִיקַת הַלֵב, אי ספיקת כליות או שבץ. תרופות כמו פניטואין וקרבמזפין ניתנות לעיתים קרובות לטיפול בכאבים המלווים את מחלת פאברי, אך לא כדי לרפא אותו. Metoclopramide או Lipisorb (תוסף תזונה) עשויים להקל על הפרעות במערכת העיכול מתרחשת לעיתים קרובות אצל אנשים הסובלים ממחלת פאברי, ואנשים מסוימים עשויים להזדקק להשתלת כליה או דיאליזה. החלפת אנזים יכולה להפחית את האחסון, להקל על הכאבים ולשמר את תפקוד האיברים אצל אנשים הסובלים ממחלת פאברי.
מחלת פרבר ידוע גם כ ליפוגרנולומטוזיס של פרבר, מתאר קבוצת הפרעות רצסיביות אוטוזומליות נדירות הגורמות להצטברות חומרים שומניים במפרקים, ברקמות ובמערכת העצבים המרכזית. זה משפיע על גברים ונשים כאחד. המחלה מתחילה בדרך כלל בגיל הרך, אך יכולה להופיע מאוחר יותר בחיים.
ילדים עם מחלת פרבר קלאסית מתפתחים סימפטומים נוירולוגיים, שעשויים לכלול ישנוניות מוגברת ועייפות, ובעיות עם בְּלִיעָה. הכבד, הלב והכליות יכולים להיות מושפעים גם כן. תסמינים אחרים עשויים לכלול התכווצויות מפרקים (קיצור כרוני של שרירים או גידים סביב המפרקים), הקאות, דלקת פרקים, הגדלה בלוטות לימפה, דלקת מפרקים, צרידות וגושים מתחת לעור המתעבים סביב המפרקים ככל שהוא מתקדם מחלות.
אנשים הסובלים מקשיי נשימה עשויים להזדקק להכנסת צינור נשימה. רוב הילדים הסובלים ממצב זה מתים עד גיל שנתיים, בדרך כלל מגיל מחלות ריאה. באחת הצורות החמורות ביותר של המחלה, ניתן לאבחן כבד וטחול מוגדל זמן קצר לאחר הלידה. תינוקות שנולדו עם סוג זה של המחלה בדרך כלל מתים תוך 6 חודשים.
מחלת פרבר נגרמת על ידי מחסור באנזים הנקרא סרמידאז. כיום אין טיפול ספציפי למחלת פרבר. ניתן לרשום קורטיקוסטרואידים להקלה על הכאבים. השתלת מח עצם יכולה להפחית גרנולומות (המונים קטנים של רקמות דלקתיות) אצל אנשים עם דלקת קלה או בחולים ללא סיבוכים ריאתיים או מערכת העצבים. אצל אנשים מבוגרים, גרנולומות יכולות להיכווץ או להסיר בניתוח.
גנגליוסידוזיס מורכב משתי קבוצות שונות של מחלות גנטיות. שניהם אוטוזומליים רצסיביים ומשפיעים על זכרים ונקבות באופן שווה.
Gangliosidosis GM1.
Gangliosidosis GM1 נגרמת על ידי מחסור באנזים בטא-גלקטוסידאז, וכתוצאה מכך הצטברות לא תקינה של חומרים ליפידים חומציים, בפרט בתאי עצב בעצב המרכזי והפריפרי מערכות. ל- Gangliosidosis GM1 יש שלושה ביטויים קליניים:
- GM1 (תת הסוג החמור ביותר, המתרחש זמן קצר לאחר הלידה) עשוי לכלול ניוון עצבי, התקפים, כבד מוגדל וטחול, גסות תווי הפנים, אי סדרים בשלד, נוקשות במפרק, נפיחות, חולשת שרירים, תגובה מבוהלת מוגזמת ובעיות עם הליכה. כמחצית מהנפגעים מפתחים כתמים אדומים בדובדבן בעין. ילדים עשויים להיות חירשים ועיוורים עד גיל 1 ולעתים קרובות מתים עד גיל 3 מסיבוכי לב או דלקת ריאות.
- ילד מאוחר Gangliosidosis GM1 בדרך כלל מתחיל בין הגילאים 1 עד 3 שנים. התסמינים הנוירולוגיים כוללים אטקסיה, התקפים, דמנציה וקושי בדיבור.
- Gangliosidosis GM1 מתפתח בין הגילאים 3 ל -30 שנים. התסמינים כוללים ירידה במסת השריר (ניוון שרירים), סיבוכים נוירולוגיים פחות חמורים ומתקדמים לאט יותר מצורות אחרות הפרעות, אטימות הקרנית אצל אנשים מסוימים והתכווצויות שרירים מתמשכות הגורמות לתנועות מתפתלות וחוזרות על עצמן או תנוחות חריגות. (דיסטוניה). אנגיוקראטומות יכולות להתפתח בגוף התחתון של הגוף. גודל הכבד והטחול תקין אצל רוב האנשים שנפגעו.
Gangliosidosis GM2.
Gangliosidosis GM2 גורם גם לגוף לצבור עודף חומרים שומניים חומציים ברקמות ובתאים, בעיקר תאי עצב. הפרעות אלו נובעות ממחסור באנזים בטא-הקסוסמינידאז. הפרעות GM2 כוללות:
- מחלת טיי זאקס (המכונה גם גרסה B2 של Gangliosidosis B) וצורות הגרסא שלה נגרמות על ידי מחסור באנזים hexosaminidase A. ילדים מושפעים מתפתחים כרגיל במהלך החודשים הראשונים לחיים. התסמינים מתחילים בגיל 6 חודשים וכוללים אובדן הדרגתי של יכולת נפשית, דמנציה, ירידה בקשר עין, עלייה תגובה מבהילה לרעש, אובדן שמיעה פרוגרסיבי המוביל לחירשות, קשיי בליעה, עיוורון, כתמים אדומים של הרשתית, וכמה שיתוק. התקפים יכולים להתחיל בשנה השנייה לחיי הילד. תינוקות עלולים בסופו של דבר להיות תלויים בצינור האכלה, ולעתים קרובות מתים עד גיל 4 כתוצאה מזיהומים שחוזרים על עצמם. למרבה הצער, אין טיפול מיוחד. תרופות נוגדות פרכוסים עשויות לשלוט בתחילה בהתקפים. טיפולים תומכים אחרים כוללים תזונה נכונה והידרציה, ושיטות לשמירה על דרכי הנשימה פתוחות. צורה נדירה של ההפרעה הנקראת מחלת טיי זאקס המאוחרת, מופיע אצל אנשים בגילאי 20 עד 30 ומאופיין בהליכה לא יציבה ובהידרדרות נוירולוגית מתקדמת.
- מחלת סנדהוף (אופציה AB) היא צורה חמורה של מחלת טיי-סאקס. ההתחלה מתרחשת בדרך כלל בגיל 6 חודשים ואינה מוגבלת לאף קבוצה אתנית. סימנים נוירולוגיים עשויים לכלול הידרדרות הדרגתית של מערכת העצבים המרכזית, מוטורית חולשה, עיוורון מוקדם, תגובה מבוהלת לקול, ספסטיות, הלם או עוויתות שרירים (מיוקלונוס), אֶפִּילֶפּסִיָה, ראש מוגדל באופן חריג (מקרוצפליה), וכתמים אדומים בעין. תסמינים אחרים עשויים לכלול דלקות בדרכי הנשימה תכופות, רחש לב, תווי פנים דמויי בובה והגדלת הכבד והטחול. אין טיפול ספציפי למחלת סנדהוף. בדומה למחלת טיי-זאקס, טיפול תומך כולל שמירה על דרכי הנשימה פתוחות כל הזמן, כמו גם תזונה נכונה והידרציה. תרופות נוגדות פרכוסים יכולות לשלוט בתחילה באפילפסיה (התקפים).
קראו גם:תסחיף מי שפיר
מחלת קראב (ידוע גם כ לוקודיסטרופיה של תאים כדוריים ו ליפידוזיס של גלקטוזילצרמיד) היא מחלה אוטוזומלית רצסיבית הנגרמת על ידי מחסור באנזים galactocerebrosidase. המחלה פוגעת לרוב בתינוקות עם הופעה לפני גיל 6 חודשים, אך יכולה להופיע במהלך גיל ההתבגרות או הבגרות.
הצטברות שומן בלתי מעוכל משפיעה על צמיחת מעטפת המגן של העצב (מעטפת המיאלין) וגורמת לפגיעה קשה במיומנויות המנטליות והמוטוריות. תסמינים אחרים כוללים חולשת שרירים, ירידה ביכולת השרירים להתמתח (היפוטוניה), מתח שרירים (ספסטיות), הלם פתאומי או עוויתות בגפיים (התקפים מיוקלוניים), עצבנות, חום בלתי מוסבר, חירשות, עיוורון, שיתוק וקשיי בליעה. ירידה במשקל לטווח ארוך יכולה להתרחש גם כן.
ניתן לאבחן את המחלה על ידי בדיקת אנזים וזיהוי קיבוץ התאים האופייני לה לגופים גלובוידיים בחומר הלבן של המוח, דמיאלינציה של עצבים והתנוונות והרס תאי המוח. אצל תינוקות המחלה בדרך כלל קטלנית לפני גיל שנתיים. לאנשים עם צורה מתקדמת יותר של המחלה יש מהלך מתון יותר של המחלה וחיים זמן רב יותר.
לא פותח טיפול ספציפי למחלת קראב, אם כי השתלת מח עצם מוקדמת עשויה לסייע לאנשים מסוימים. לאנשים עם צורה מתקדמת יותר של המחלה יש מהלך מתון יותר של המחלה וחיים זמן רב יותר.
לוקודיסטרופיה מטכרומטית, או MLD, היא קבוצת הפרעות המסומנות על ידי הצטברות חומרים שומניים בחומר הלבן של מערכת העצבים המרכזית ובעצבים היקפיים ובמידה מסוימת בכליות. בדומה למחלת קראב, MLD משפיע על המיאלין, המכסה ומגן על העצבים. הפרעה אוטוזומלית רצסיבית זו נגרמת על ידי מחסור באנזים arylsulfatase A. הפרעה זו משפיעה על גברים ונשים כאחד.
ללוקודיסטרופיה מטכרומטית יש שלוש צורות אופייניות: תינוק מאוחר, צעיר ומבוגר.
- MLD לתינוקות מאוחרים בדרך כלל מתחיל בין 12 ל -20 חודשים לאחר הלידה. ילדים עשויים להיראות רגילים בהתחלה, אך מתקשים בהליכה ונטייה ליפול, מלווה בכאבים לסירוגין בידיים וברגליים. אובדן ראייה פרוגרסיבי המוביל לעיוורון, עיכובים התפתחותיים ואובדן אבני דרך שנרכשו בעבר, ליקוי בליעה, התקפים ודמנציה עד 2 שנים. ילדים מפתחים גם בזבוז שרירים וחולשה מתקדמים ובסופו של דבר מאבדים את כושר ההליכה. רוב הילדים הסובלים מהפרעה זו מתים עד גיל 5.
- MLD לנוער בדרך כלל מתחיל מגיל 3 עד 10 שנים. התסמינים כוללים פגיעה בביצועי בית הספר, פגיעה נפשית, אטקסיה, התקפים ודמנציה. הסימפטומים מתקדמים, כאשר המוות מתרחש 10-20 שנים לאחר הופעת המחלה.
- צורת מבוגרים. תסמינים מבוגרים הופעה לאחר גיל 16 ועשויה לכלול אטקסיה, התקפים, רעידות חריגות בגפיים (רעידות), פגיעה בריכוז, דִכָּאוֹן, הפרעות נפשיות ודמנציה. בדרך כלל המוות מתרחש 6-14 שנים לאחר הופעת התסמינים.
לא ניתן לרפא MLD. הטיפול סימפטומטי ותומך. במקרים מסוימים השתלת מח עצם יכולה לעכב את התקדמות המחלה. התקדמות משמעותית חלה בטיפול גנטי במודלים של בעלי חיים עם MLD ובניסויים קליניים.
מחלת וולמןידוע גם כ חסר ליפוז חומצה ליזוזומליתהיא הפרעה חמורה באגירת שומנים שהיא בדרך כלל קטלנית בגיל שנה. הפרעה אוטוזומלית רצסיבית זו מאופיינת בהצטברות של אסטרי כולסטרול (בדרך כלל צורת ההובלה של הכולסטרול) ו טריגליצרידים (הצורה הכימית שבה קיימים שומנים בגוף), שיכולים להצטבר באופן משמעותי ולגרום לנזק לתאים ו בדים.
גברים ונשים כאחד סובלים מהפרעה זו. תינוקות רגילים ופעילים בלידה, אך מפתחים במהירות פגיעה נפשית מתקדמת, כבד מוגדל וטחול מוגדל מאוד, נפיחות, בעיות במערכת העיכול. צַהֶבֶת, אנמיה, הקאות ופיקדונות סידן ב בלוטות יותרת הכליהגורם להם להתקשות.
סוג אחר מחסור ליפוז חומצה ליזוזומלית הוא מחלת אחסון כולסטרול אסטרס. מחלה נדירה ביותר זו מתרחשת כתוצאה מאחסון אסטרי כולסטרול ו טריגליצרידים בתאי הדם, לימפה ורקמת לימפה. בילדים בבגרות הכבד מתרחב, מה שמוביל לשחמת ו כְּרוֹנִי כשל בכבד. לילדים עשויים להיות גם מצבורי סידן בבלוטת יותרת הכליה, וצהבת עלולה להתפתח בשלבים המאוחרים של המחלה.
נכון לעכשיו, נושא החלפת האנזים נחקר באופן פעיל הן במחלת וולמן והן בהצטברות אסטר כולסטרול.


