
Lipoidoze (lizosomu lipīdu uzkrāšanās slimība): kas tas ir, piemēri
Uzmanību! Informācija ir paredzēta tikai informatīviem nolūkiem, un tā nav paredzēta diagnozes noteikšanai un ārstēšanas izrakstīšanai. Vienmēr konsultējieties ar savu ārstu-speciālistu!
Saturs
- Kas ir lizosomu uzkrāšanās slimības?
- Kas ir lipīdi?
- Kā lipoidoze tiek mantota?
- Lizosomu uzkrāšanās slimību veidi un simptomi
Kas ir lizosomu uzkrāšanās slimības?
Lizosomu uzkrāšanās slimības (lipoidoze), ir iedzimtu vielmaiņas slimību grupa, kurā dažādās ķermeņa šūnās un audos uzkrājas kaitīgs daudzums taukvielu (lipīdu).
Cilvēki ar šiem traucējumiem vai nu nepietiekami ražo kādu no enzīmiem, kas nepieciešami lipīdu sadalīšanai (metabolizācijai), vai arī viņi ražo fermentus, kas nedarbojas pareizi.
Laika gaitā šī pārmērīgā tauku uzkrāšanās var izraisīt neatgriezeniskus šūnu un audu bojājumus, īpaši smadzenēs, perifērajā nervu sistēmā (nervi no muguras smadzenēm līdz pārējām ķermenis), aknas, liesa un kaulu smadzenes.
Kas ir lipīdi?
Lipīdi ir taukiem līdzīgas vielas, kas ir svarīgas membrānu daļas šūnās un starp šūnām, kā arī mielīna apvalkā, kas pārklāj un aizsargā nervus. Lipīdi ir eļļas, taukskābes, vaski, steroīdi (piemēram, holesterīns un estrogēns) un citi saistīti savienojumi.
Šīs taukvielas dabiski uzglabājas ķermeņa šūnās, orgānos un audos. Sīkie ķermeņi šūnās, ko sauc par lizosomām, regulāri pārvērš vai metabolizē lipīdus un olbaltumvielas mazākos komponentos, lai nodrošinātu ķermenim enerģiju. Traucējumus, kuros intracelulārais materiāls, ko nevar metabolizēt, paliek lizosomās, sauc par lizosomu uzkrāšanās slimībām.
Papildus slimībām, kas saistītas ar lipīdu uzkrāšanos, citas lizosomu slimības, kas saistītas ar lipīdu uzkrāšanos, ietver mukolipidozes, kurās uzkrājas šūnas un audi. pārmērīgs lipīdu daudzums ar tiem piesaistītām cukura molekulām, kā arī mukopolisaharidozes, kurās uzkrājas pārmērīgi daudz liela, sarežģīta cukura molekulas.
Kā tiek mantotas lipoidoze?
Lizosomu uzkrāšanās slimības tiek mantotas no viena vai abiem vecākiem, kuriem ir bojāts gēns, kas regulē noteiktu lipīdus metabolizējošo enzīmu ķermeņa šūnu klasē. Tos var mantot divos veidos:
- Autosomāli recesīvs mantojums rodas, ja abi vecāki nēsā un nodod bojātā gēna kopiju, bet traucējumi neietekmē nevienu no vecākiem. Katram bērnam, kas dzimis šiem vecākiem, ir 25 procenti iespēja mantot abas bojātā gēna kopijas, 50 procenti iespējamība, ka viņš būs pārnēsātājs, kas līdzīgs saviem vecākiem, un 25 procentu iespēja nemantot nevienu bojātā eksemplāru. gēns. Abu dzimumu bērni var tikt mantoti autosomāli recesīvā veidā.
- Ar X saistīts (vai ar dzimumu saistīts) recesīvs mantojums rodas, kad māte pārnēsā skarto gēnu X hromosomā. X un Y hromosomas ir iesaistītas dzimuma noteikšanā. Sievietēm ir divas X hromosomas, bet vīriešiem viena X hromosoma un viena Y hromosoma. Sieviešu nēsātāju dēliem ir 50 procentu iespēja mantot un ietekmēt traucējumus, jo dēli saņem vienu X hromosomu no savas mātes un vienu Y hromosomu no sava tēva. Meitām ir 50 procenti iespēja mantot skarto X hromosomu no savas mātes, un tās ir nēsātājas vai ir viegli ietekmētas. Slimie vīrieši slimību nepārnēsā saviem dēliem, bet viņu meitas būs slimības nesēji un nesēji.
Lizosomu uzkrāšanās slimību veidi un simptomi
Gošē slimība - visizplatītākā no lizosomu slimībām. Slimību izraisa enzīma glikocerebrozidāzes deficīts, kas izraisa glikocerebrozīda uzkrāšanos daudzos audos. Tauku materiāls var uzkrāties smadzenēs, liesā, aknās, nierēs, plaušās un kaulu smadzenēs.
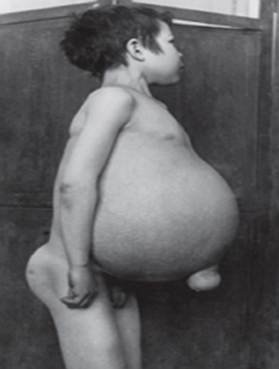
Simptomi var ietvert smadzeņu bojājumus, liesas palielināšanās un aknas, aknu darbības traucējumi, skeleta traucējumi un kaulu bojājumi, kas var izraisīt sāpes un lūzumus, limfmezglu un (dažreiz) blakus esošo locītavu pietūkumu, vēdera uzpūšanās, brūngans ādas tonis, anēmija, zems trombocītu skaits un dzelteni plankumi uz acīm.
Visnopietnāk skartās personas var būt arī jutīgākas pret infekcijām. Slimība vienādi skar vīriešus un sievietes.
Gošē slimībai ir trīs vispārīgi klīniskie apakštipi:
- 1. veids (Vai nē neironopātisks veids) ir visizplatītākā slimības forma Amerikas Savienotajās Valstīs un Eiropā. Smadzenes netiek ietekmētas, taču var būt plaušu un retos gadījumos arī nieru bojājumi. Simptomi var sākties agrīnā dzīves posmā vai pieaugušā vecumā, un tie ietver aknu palielināšanos un smagu liesas palielināšanos, kas var izraisīt plīsumu un papildu komplikācijas. Skeleta vājums un kaulu slimības var būt plaši izplatītas. Šīs grupas cilvēkiem parasti ir viegli zilumi, jo viņu asinīs ir zems trombocītu skaits. Viņiem var būt arī nogurums anēmijas dēļ. Atkarībā no slimības sākuma un smaguma pakāpes cilvēki ar 1. veids var nodzīvot līdz pilngadībai. Daudziem slimniekiem ir viegla slimība, vai arī viņiem var nebūt nekādu simptomu. Lai gan 1veids Gošē slimība ir izplatīta Ashkenazi ebreju mantojuma cilvēku vidū, un tā var skart dažādu etnisko piederību cilvēkus.
- 2. veids (vai akūta bērnības neiropātija Gošē slimība) parasti sākas 3 mēnešu laikā pēc dzimšanas. Simptomi ir plaši un progresējoši smadzeņu bojājumi, spasticitāte, krampji, stīvums ekstremitātēs, palielinātas aknas (aknu hepatomegālija) un liesa (splenomegālija), patoloģiskas acu kustības un slikta sūkšanas un norīšanas spēja. Slimie bērni parasti mirst pirms 2 gadu vecuma.
- 3. veids (hroniska neironopātiska forma) var sākties jebkurā bērnībā vai pat pieaugušā vecumā. To raksturo lēni progresējoši, bet mazāk smagi neiroloģiski simptomi, salīdzinot ar akūtu Gošē slimību 2 veidi. Galvenie simptomi ir acu kustību traucējumi, kognitīvie traucējumi, slikta koordinācija, krampji, liesas un/vai aknu palielināšanās, skeleta slimības, asins slimības, tostarp anēmija, un problēmas ar elpošana. Gandrīz visi ar Gošē slimību 3 veidi, kurš saņem enzīmu aizstājterapiju, sasniegs pilngadību.
Pacientiem 1. un 3. tips Enzīmu aizstājterapija, ko ievada intravenozi ik pēc divām nedēļām, var ievērojami samazināt aknu un liesas izmēru, samazināt skeleta anomālijas un novērst citas izpausmes. Veiksmīga kaulu smadzeņu transplantācija ārstē slimības neiroloģiskās izpausmes. Tomēr šī procedūra ir saistīta ar ievērojamu risku un reti tiek veikta personām ar Gošē slimību.
Retos gadījumos var būt nepieciešama operācija, lai noņemtu visu liesu vai tās daļu (ja cilvēkam ir ļoti zems trombocītu skaits vai ja palielināts orgāns būtiski ietekmē cilvēka komfortu). Asins pārliešana var būt noderīga dažiem cilvēkiem ar anēmiju. Citiem var būt nepieciešama locītavu protezēšanas operācija, lai uzlabotu mobilitāti un dzīves kvalitāti. Pašlaik nav efektīvas ārstēšanas smadzeņu bojājumiem, kas var rasties cilvēkiem ar 2. un 3. tipa Gošē slimību.
Nīmaņa-Pika slimība ir autosomāli recesīvu traucējumu grupa, ko izraisa tauku un holesterīna uzkrāšanās aknu, liesas, kaulu smadzeņu, plaušu un dažos gadījumos arī smadzeņu šūnās.
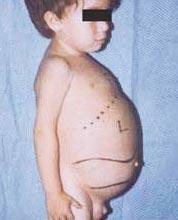
Neiroloģiskas komplikācijas var būt ataksija (muskuļu koordinācijas trūkums, kas var ietekmēt staigāšanu, rakstīšanu un ēšanu, cita starpā), acu paralīze, smadzeņu deģenerācija, mācīšanās problēmas, spasticitāte, apgrūtināta barošana un rīšana, neskaidra runa, muskuļu tonusa zudums, paaugstināta jutība pret pieskārienu un radzenes apduļķošanās pārmērīgas uzkrāšanās dēļ materiāliem. 50% skarto cilvēku ap tīklenes centru veidojas raksturīgs ķiršu sarkans oreols, ko ārsts var redzēt ar īpašu instrumentu.
Nīmaņa-Pika slimību iedala trīs vispārīgos apakštipos:
- A tips, vissmagākā forma, sākas agrā bērnībā. Zīdaiņi pēc piedzimšanas izskatās normāli, bet līdz 6 mēnešiem attīstās dziļi smadzeņu bojājumi, palielinās aknas un liesa, kā arī pietūkuši limfmezgli un zemādas mezgli (ksantomas). Liesa var kļūt 10 reizes lielāka par parasto izmēru un var plīst, izraisot asiņošanu. Šie bērni kļūst arvien vājāki, zaudē motoriskās funkcijas, var kļūt anēmiski un ir pakļauti atkārtotām infekcijām. Viņi reti dzīvo ilgāk par 18 mēnešiem. Šī slimības forma visbiežāk sastopama ebreju ģimenēs.
- B tips(vai nepilngadīgo sākums) parasti neietekmē smadzenes, bet lielākā daļa bērnu attīstās ataksija, nervu bojājumi, kas atstāj muguras smadzenes (perifēra neiropātija), un plaušu problēmas, kas progresē līdz ar vecumu. Pusaudžiem raksturīga aknu un liesas palielināšanās. Cilvēki ar B tips var dzīvot salīdzinoši ilgi, bet daudziem nepieciešama mūža skābekļa terapija plaušu bojājumu dēļ. Niemann-Pick A un B tips ir taukainas vielas, ko sauc par sfingomielīnu, uzkrāšanās rezultāts enzīma, ko sauc par sfingomielināzi, deficīta dēļ.
- C tips var parādīties agrīnā dzīves posmā vai attīstīties pusaudža vai pat pieaugušā vecumā. Nīmaņa-Pika slimībaC tips izraisa nevis sflingomielināzes deficīts, bet gan NPC1 vai NPC2 proteīnu trūkums. Rezultātā dažādi lipīdi un īpaši holesterīns uzkrājas nervu šūnās un izraisa to darbības traucējumus. Smadzeņu iesaistīšanās var būt plaša, izraisot nespēju skatīties uz augšu un uz leju, apgrūtinātu staigāšanu un rīšanu, progresējošu dzirdes zudumu un progresējošu. demenci. Cilvēki ar C tips ir tikai neliela liesas un aknu palielināšanās. Cilvēku ar C tips ievērojami atšķiras. Daži cilvēki mirst bērnībā, savukārt citi, kas ir mazāk skarti, var dzīvot arī pieaugušā vecumā.
Pašlaik Nīmaņa-Pika slimību nevar izārstēt. Ārstēšana ir atbalstoša. Bērni parasti mirst no infekcijas vai progresējoša neiroloģiska zaudējuma. Ir bijuši kaulu smadzeņu transplantācijas gadījumi vairākiem pacientiem ar B tips ar jauktiem rezultātiem.
Fabri slimībazināms arī kā alfa-galaktozidāzes-A deficīts, izraisa taukainu vielu uzkrāšanos autonomajā nervu sistēmā (tajā nervu sistēmas daļā, kas kontrolē tādas piespiedu funkcijas kā elpošana un sirdsdarbība), acīs, nierēs un sirds un asinsvadu sistēmā sistēma.
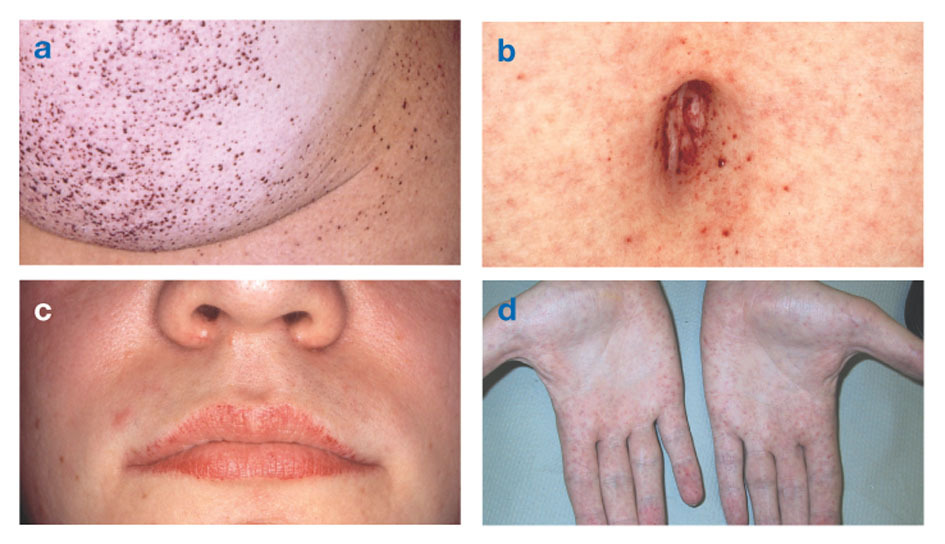
Fabri slimība ir vienīgā ar X saistītā lipīdu slimība. Šī slimība galvenokārt skar vīriešus, lai gan sievietēm tā ir vieglāka un daudzveidīgāka. Reti skartajām sievietēm ir smagi simptomi, kas līdzīgi vīriešiem ar šo traucējumu. Simptomi parasti parādās bērnībā vai pusaudža gados.
Neiroloģiskas pazīmes ietver dedzinošas sāpes rokās un kājās, kas pastiprinās karstā laikā vai pēc tam vingrošana, kā arī liekā materiāla uzkrāšanās radzenes caurspīdīgajos slāņos (kas izraisa duļķošanos, bet bez redzes izmaiņas). Tauku uzkrāšanās asinsvadu sieniņās var traucēt asinsriti, pakļaujot cilvēku riskam insults vai sirdstrieka. Citi simptomi ir palielināta sirds, progresējoša nieru mazspējaizraisot nieru slimību beigu stadijā, kuņģa-zarnu trakta problēmas, samazinātu svīšanu un drudzi.
Angiokeratomas (mazi, bezvēža, sarkanīgi purpursarkani, pacelti plankumi uz ādas) var veidoties rumpja lejasdaļā un kļūt biežāki ar vecumu.
Cilvēki ar Fābri slimību bieži mirst priekšlaicīgi no komplikācijām no sirdskaite, nieru mazspēja vai insults. Tādas zāles kā fenitoīns un karbamazepīns bieži tiek parakstītas, lai ārstētu sāpes, kas pavada Fābri slimību, bet ne, lai tās izārstētu. Metoklopramīds vai Lipisorb (uztura bagātinātājs) var mazināt kuņģa-zarnu trakta traucējumus bieži rodas cilvēkiem ar Fābri slimību, un dažiem cilvēkiem var būt nepieciešama nieres transplantācija vai dialīze. Fermentu aizstāšana dažiem cilvēkiem ar Fābri slimību var samazināt uzglabāšanu, mazināt sāpes un saglabāt orgānu darbību.
Farbera slimība zināms arī kā Farbera lipogranulomatoze, apraksta retu autosomāli recesīvu traucējumu grupu, kas izraisa taukainu vielu uzkrāšanos locītavās, audos un centrālajā nervu sistēmā. Tas skar gan vīriešus, gan sievietes. Parasti slimība sākas agrā bērnībā, bet var rasties arī vēlāk.
Attīstās bērni ar klasisko Farbera slimību neiroloģiski simptomi, kas var ietvert pastiprinātu miegainību un letarģiju, kā arī problēmas ar norijot. Var tikt ietekmētas arī aknas, sirds un nieres. Citi simptomi var būt locītavu kontraktūras (hroniska muskuļu vai cīpslu saīsināšanās ap locītavām), vemšana, artrīts, paplašināšanās. limfmezgli, locītavu iekaisums, aizsmakums un zemādas kunkuļi, kas sabiezē ap locītavām progresējot slimības.
Skartajiem cilvēkiem ar apgrūtinātu elpošanu var būt nepieciešams ievietot elpošanas cauruli. Lielākā daļa bērnu ar šo stāvokli mirst līdz 2 gadu vecumam, parasti no plaušu slimības. Vienā no smagākajām slimības formām īsi pēc piedzimšanas var diagnosticēt palielinātas aknas un liesu. Zīdaiņi, kas dzimuši ar šo slimības formu, parasti mirst 6 mēnešu laikā.
Farbera slimību izraisa enzīma, ko sauc par keramidāzi, deficīts. Pašlaik Farbera slimībai nav specifiskas ārstēšanas. Sāpju mazināšanai var ordinēt kortikosteroīdus. Kaulu smadzeņu transplantācija var samazināt granulomas (nelielas iekaisušo audu masas) cilvēkiem ar vieglu iekaisumu vai pacientiem bez plaušu vai nervu sistēmas komplikācijām. Gados vecākiem cilvēkiem granulomas var samazināt vai ķirurģiski noņemt.
Gangliozidoze sastāv no divām dažādām ģenētisko slimību grupām. Abi ir autosomāli recesīvi un vienādi ietekmē vīriešus un sievietes.
GM1 gangliozidoze.
GM1 gangliozidozi izraisa enzīma beta-galaktozidāzes deficīts, kā rezultātā rodas patoloģiska skābie lipīdu materiāli, jo īpaši centrālās un perifērās nervu šūnās sistēmas. GM1 gangliozidozei ir trīs klīniskas izpausmes:
- GM1 (visnopietnākais apakštips, kas rodas drīz pēc dzimšanas) var ietvert neirodeģenerāciju, krampjus, palielinātas aknas un liesu, sejas vaibstu rupjība, skeleta nelīdzenumi, locītavu stīvums, vēdera uzpūšanās, muskuļu vājums, pārspīlēta reakcija uz pārsteigumu un problēmas ar gaita. Apmēram pusei no skartajām acīs veidojas ķiršu sarkani plankumi. Bērni var būt kurli un akli līdz 1 gada vecumam un bieži mirst līdz 3 gadu vecumam no sirds komplikācijām vai pneimonija.
- Vēls bērns GM1 gangliozidoze parasti sākas vecumā no 1 līdz 3 gadiem. Neiroloģiskie simptomi ir ataksija, krampji, demence un runas grūtības.
- GM1 gangliozidoze attīstās vecumā no 3 līdz 30 gadiem. Simptomi ir samazināta muskuļu masa (muskuļu atrofija), neiroloģiskas komplikācijas, kas ir mazāk smagas un progresē lēnāk nekā citas formas. traucējumi, radzenes apduļķošanās dažiem cilvēkiem un ilgstošas muskuļu kontrakcijas, kas izraisa griešanos un atkārtotas kustības vai neparastas pozas (distonija). Angiokeratomas var attīstīties ķermeņa lejasdaļā. Lielākajai daļai skarto cilvēku aknu un liesas izmērs ir normāls.
GM2 gangliozidoze.
GM2 gangliozidoze izraisa arī pārmērīgu skābo taukskābju vielu uzkrāšanos audos un šūnās, galvenokārt nervu šūnās. Šos traucējumus izraisa beta-heksosaminidāzes enzīma deficīts. GM2 traucējumi ietver:
- Tay-Sachs slimība (pazīstams arī kā GM2 gangliozidozes B variants) un tā variantu formas izraisa enzīma heksosaminidāzes A deficīts. Skartie bērni pirmajos dzīves mēnešos attīstās normāli. Simptomi sākas 6 mēnešu vecumā un ietver progresējošu garīgo spēju zudumu, demenci, samazinātu acu kontaktu, palielinātu satriecoša reakcija uz troksni, progresējošs dzirdes zudums, kas izraisa kurlumu, apgrūtināta rīšana, aklums, ķiršu sarkani plankumi uz tīklenes un daži paralīze. Krampji var sākties bērna otrajā dzīves gadā. Zīdaiņi galu galā var kļūt atkarīgi no barošanas caurules un bieži mirst līdz 4 gadu vecumam no atkārtotām infekcijām. Diemžēl īpašas ārstēšanas nav. Pretkrampju līdzekļi sākotnēji var kontrolēt krampjus. Citas atbalstošas ārstēšanas metodes ietver pareizu uzturu un hidratāciju, kā arī metodes elpceļu atvēršanai. Reta traucējuma forma, ko sauc novēlota Tay-Sachs slimība, rodas cilvēkiem vecumā no 20 līdz 30 gadiem, un to raksturo nestabila gaita un progresējoša neiroloģiska stāvokļa pasliktināšanās.
- Sandhofa slimība (AB variants) ir smaga Tay-Sachs slimības forma. Sākums parasti notiek 6 mēnešu vecumā un nav ierobežots nevienai etniskajai grupai. Neiroloģiskas pazīmes var ietvert progresējošu centrālās nervu sistēmas, motora pasliktināšanos vājums, agrs aklums, smaga izbijusies reakcija uz skaņu, spasticitāte, šoks vai raustīšanās muskuļi (mioklonuss), epilepsija, patoloģiski palielināta galva (makrocefālija) un sarkani plankumi acī. Citi simptomi var ietvert biežas elpceļu infekcijas, sirds trokšņi, lellei līdzīgi sejas vaibsti un aknu un liesas palielināšanās. Sandhofa slimībai nav specifiskas ārstēšanas. Tāpat kā Tay-Sachs slimības gadījumā, atbalstošā aprūpe ietver elpceļu nepārtrauktu atvēršanu, kā arī pareizu uzturu un hidratāciju. Pretkrampju līdzekļi sākotnēji var kontrolēt epilepsiju (krampjus).
Krabbes slimība(zināms arī kā lodveida šūnu leikodistrofija un galaktozilkeramīda lipidoze) ir autosomāli recesīva slimība, ko izraisa enzīma galaktocerebrozidāzes deficīts. Visbiežāk šī slimība skar zīdaiņus pirms 6 mēnešu vecuma, bet var rasties pusaudža vai pieaugušā vecumā.
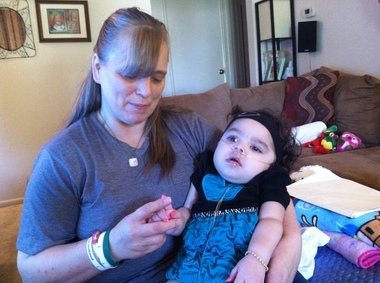
Nesagremoto tauku uzkrāšanās ietekmē nervu aizsargapvalka (mielīna apvalka) augšanu un izraisa nopietnus garīgo un motorisko prasmju traucējumus. Citi simptomi ir muskuļu vājums, samazināta muskuļu spēja stiepties (hipotonija), muskuļu sasprindzinājums (spastiskums), pēkšņs šoks vai ekstremitāšu raustīšanās (miokloniski krampji), aizkaitināmība, neizskaidrojams drudzis, kurlums, aklums, paralīze un apgrūtināta rīšana. Var rasties arī ilgtermiņa svara zudums.
Slimību var diagnosticēt, pārbaudot enzīmus un nosakot tai raksturīgo šūnu grupu globoīdos ķermeņos smadzeņu baltajā vielā, nervu demielinizācija un deģenerācija, kā arī smadzeņu šūnu iznīcināšana. Zīdaiņiem slimība parasti ir letāla pirms 2 gadu vecuma. Cilvēkiem ar progresīvāku slimības formu slimības gaita ir vieglāka un dzīvo ievērojami ilgāk.
Nav izstrādāta specifiska Krabbes slimības ārstēšana, lai gan agrīna kaulu smadzeņu transplantācija var palīdzēt dažiem cilvēkiem. Cilvēkiem ar progresīvāku slimības formu slimības gaita ir vieglāka un dzīvo ievērojami ilgāk.
Metahromatiskā leikodistrofija, jeb MLD, ir traucējumu grupa, ko raksturo taukainu vielu uzkrāšanās centrālās nervu sistēmas baltajā vielā un perifērajos nervos un zināmā mērā nierēs. Līdzīgi kā Krabbes slimība, MLD ietekmē mielīnu, kas pārklāj un aizsargā nervus. Šo autosomālo recesīvo traucējumu izraisa enzīma arilsulfatāzes A deficīts. Šis traucējums skar gan vīriešus, gan sievietes.
Metahromatiskajai leikodistrofijai ir trīs raksturīgas formas: vēls zīdainis, nepilngadīgais un pieaugušais.
- Vēlīnā zīdaiņa MLD parasti sākas no 12 līdz 20 mēnešiem pēc dzimšanas. Bērni sākumā var izskatīties normāli, bet viņiem ir grūtības staigāt un tendence krist, ko pavada periodiskas sāpes rokās un kājās. progresējošs redzes zudums, kas izraisa aklumu, attīstības aizkavēšanos un iepriekš iegūto atskaites punktu zudumu, rīšanas traucējumus, krampjus un demenci līdz 2 gadiem. Bērniem arī attīstās progresējoša muskuļu izsīkšana un vājums, un galu galā viņi zaudē spēju staigāt. Lielākā daļa bērnu ar šo traucējumu formu mirst līdz 5 gadu vecumam.
- Nepilngadīgo MLD parasti sākas vecumā no 3 līdz 10 gadiem. Simptomi ir traucēta veiktspēja skolā, garīgi traucējumi, ataksija, krampji un demence. Simptomi progresē, nāve iestājas 10–20 gadus pēc slimības sākuma.
- Pieaugušā forma. Simptomi pieaugušie sākas pēc 16 gadu vecuma un var ietvert ataksiju, krampjus, patoloģisku ekstremitāšu trīci (trīci), koncentrēšanās traucējumus, depresija, garīgi traucējumi un demence. Nāve parasti iestājas 6–14 gadus pēc simptomu parādīšanās.
MLD nevar izārstēt. Ārstēšana ir simptomātiska un atbalstoša. Kaulu smadzeņu transplantācija dažos gadījumos var aizkavēt slimības progresēšanu. Ievērojams progress ir panākts ar gēnu terapiju dzīvnieku modeļos ar MLD un klīniskajos pētījumos.
Volmena slimībazināms arī kā lizosomu skābes lipāzes deficītsir nopietns lipīdu uzkrāšanās traucējums, kas parasti ir letāls 1 gada vecumā. Šo autosomālo recesīvo traucējumu raksturo holesterīna esteru (parasti holesterīna transporta forma) uzkrāšanās un triglicerīdi (ķīmiskā forma, kādā organismā pastāv tauki), kas var ievērojami uzkrāties un izraisīt šūnu bojājumus un audumi.
Gan vīrieši, gan sievietes cieš no šīs slimības. Zīdaiņi dzimšanas brīdī ir normāli un aktīvi, bet strauji attīstās progresējoši garīgi traucējumi, palielinās aknas un ievērojami palielināta liesa, vēdera uzpūšanās, kuņģa-zarnu trakta problēmas. dzelte, anēmija, vemšana un kalcija nogulsnes virsnieru dziedzeriizraisot to sacietēšanu.
Cits veids lizosomu skābes lipāzes deficīts ir holesterīna estera uzglabāšanas slimība. Šī ārkārtīgi retā slimība rodas holesterīna esteru uzkrāšanās rezultātā un triglicerīdi asins šūnās, limfas un limfoīdo audu. Bērniem pieaugušā vecumā palielinās aknas, kas izraisa cirozi un hroniska aknu mazspēja. Bērniem var būt arī kalcija nogulsnes virsnieru dziedzeros, un vēlākās slimības stadijās var attīstīties dzelte.
Šobrīd jautājums par enzīmu aizstāšanu tiek aktīvi pētīts gan Volmena slimībā, gan holesterīna estera akumulācijā.
Uzmanību! Informācija ir paredzēta tikai informatīviem nolūkiem, un tā nav paredzēta diagnozes noteikšanai un ārstēšanas izrakstīšanai. Vienmēr konsultējieties ar savu ārstu-speciālistu!