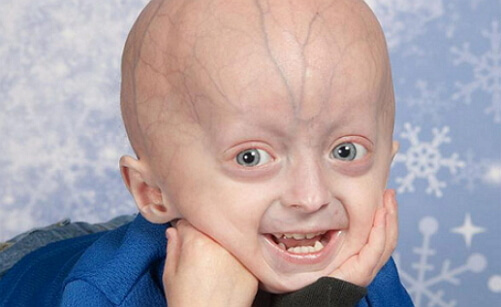
Prader-Willi sindroms
 Prādera-Willi sindroms - ģenētiska reta anomālija kas raksturīgs ar darbību gēnu apstāšanās, vai zinātniskā veidā, šī parādība, ko izraisa trūkums ekspresijas kas nozīmē nodošanu ģenētiskās informācijas no DNS RNS.Ģenētisko funkciju šajā gadījumā ir iekļaut darbā pienācīgā laikā.Piemēram, zēniem bērnībā nav matu augšanas sejas, un šī parādība sākas pēc pubertātes sākuma.Šo slimību vispirms aprakstīja 1956. gadā.
Prādera-Willi sindroms - ģenētiska reta anomālija kas raksturīgs ar darbību gēnu apstāšanās, vai zinātniskā veidā, šī parādība, ko izraisa trūkums ekspresijas kas nozīmē nodošanu ģenētiskās informācijas no DNS RNS.Ģenētisko funkciju šajā gadījumā ir iekļaut darbā pienācīgā laikā.Piemēram, zēniem bērnībā nav matu augšanas sejas, un šī parādība sākas pēc pubertātes sākuma.Šo slimību vispirms aprakstīja 1956. gadā.
Prader-Willy sindroms sastopams ar 12 000 bērnu piedzimšanas gadījumiem. Slimība ir aprakstīta darbu Haynrih Villi, Andrea Prader, Endryu Zigler, Alexis Labhart Guido Fankoni.
Prādera-Willi Syndrome izraisa
Slimība raksturo tas, ka nav vai, izsakot septiņas gēnus no piecpadsmitajā hromosomas, iedzimta no tēva. Prādera-Villi sindromu ir atzīmēta ar izmaiņām vecāku hromosomas, un gadījumā, ja mainās mātes hromosomu notiek Angelman sindromu.
Gēns komplekts, izraisot rašanos sindroma Pradera-Villija sindromu, kopiju gēns, kas atvasināts no tēva, kas aktīvi darbojas, un mātes nav klāt. Tas nozīmē, ka, ja lielākajai daļai cilvēku ir viena šo gēnu darba kopija, tad pacienti ar Pradera-Vilija sindromu dzīvo bez šādas kopijas.
Prādera-Villi sindroms simptomi
šo slimību raksturo gūžas displāziju, hipotonija( pazemināts muskuļu tonuss), aptaukošanās( fona pārēsties rodas 2. gadā).Cieš no Pradera-Villija sindromu, ir samazināts kustību koordināciju, kā arī zema kaulu blīvumu, ir īpašnieki mazo kājām un rokām, īsa auguma, slīpi gulēt, šķielēšana, ir mugurkaula deformācija.Šādiem cilvēkiem ir raksturīgas biežas siekalas, hipogonadisms( samazinātas dzimumdziedzeru funkcijas), sliktu zobu klātbūtne, neauglība. Pacientiem raksturīga garīgā atpalicība, kā arī runas attīstība, pubertātes novecošanās un grūtības apgūt mehāniskās prasmes.
Prādera-Villi sindroma un slimības pazīmes ir vizuāli novērots liels tiltu deguna, šaura un augsta piere, mandeļveida acīm, šauras lūpām. Visas šīs pazīmes un simptomi neparādās vienam pacientam. Bieži pacients sastop līdz pieciem no iepriekš minētajiem simptomiem.
Prader-Willi sindroma diagnostika
Šī slimība var tikt diagnosticēta pirms dzimšanas. To apliecina zemo mobilitāti auglim vai bērnam bieži vien ir nepareiza pozīcija, un polyhydramnios( amnija šķidrumu, kas pārmērīgā daudzumā).
Prader-Willi sindroms bieži tiek diagnosticēts pēc ģenētiskās analīzes.Šo analīzi veic jaundzimušais ar hipotonisku( samazināts muskuļu tonuss).Dažreiz ārsti pieļauj kļūdas un diagnosticē Dauna sindromu vai miopātiju. Pazīmes slimības diagnosticēšanā pēc piedzimšanas ir: dzimšanas caur ķeizargrieziena, pārkāpt prezentāciju, hipotensija, letarģijas, vāja nepieredzējis reflekss, jo samazināta muskuļu tonusa toddler, apgrūtināta elpošana, hipogonādismu.
Bērni ar Pradera-Villija sindromu, ir līdzīgi viens otram, tāpēc pieredzējis ģenētiķis uzreiz diagnosticē slimības, pat negaidot ar asins analīžu rezultātiem.
Prādera-Villi sindroma un Angelman sindroms
Šodien mēs zinām, vēl vienu slimību, kas saistītas ar māsu slimību Pradera Villija sindromu.Šo slimību sauca par Angelman. To raksturo mutācijas ģenētiskais materiāls mātei. Vairumā neatkarīgo pētījumu rezultāti norāda uz tādiem slimības cēloņiem kā mutācijas gēnu.Šī gēna produkts ir visa kompleksa olbaltumvielu degradācijas sistēmas fermentu komponents.Šī slimība ir nosaukta pēc Lielbritānijas pediatra Harija Angelmaņa, kas to raksturoja 1965. gadā.
Prādera-Willi sindromu apstrādāts
slimība, kas ir iedzimta ģenētiska anomālija netiek izzināta līdz beigām un līdzekļi ārstēšanas nav bijusi efektīva.
Kā ārstēt Prader-Willi sindromu? Tomēr ir daži medicīniski pasākumi, kas uzlabo cilvēku dzīves kvalitāti. Piemēram, bērniem ar hipotensiju ir nepieciešama masāža, kā arī cita veida īpaša terapija. Tas parāda, kā tiek izmantotas speciālās metodes, lai uzlabotu bērna attīstību, kā arī nodarbības ar defektologu un logopēdi. Bērniem 7, 8, 9 gadu vecumā ar Prader-Willi sindromu tiek piešķirti augšanas hormoni, indicēta arī hormonāla terapija ar gonadotropīniem.
Prader-Willy sindroms zēniem izpaužas kā hipogonadisms, mikrofenija un kriptorichidisms( nevis sēklinieku ovulācija).Šādā situācijā ārsti iesaka vai jāgaida, līdz sēklinieki samazinās, vai ieteiks operāciju ar hormonterapiju. Lai koriģētu svara pieaugumu, ir noteikts diētu, kas ierobežo ogļhidrātu un tauku daudzumu. Ja aptaukošanās jau ir pieejama, tad jāuzrauga pārtikas daudzums un kvalitāte, ko absorbē pacienti ar Prader-Willi sindromu.Šādiem cilvēkiem ir raksturīga vilku apetīte. Iespējama apnoja slimības gaitas komplikācija, ko raksturo elpas kavēšanās sapnī.
Pradera-Willy sindroma prognoze
Plānojot otru bērnu, šiem pašiem vecākiem var būt otrais šīs slimības risks. Tas ir atkarīgs no mehānisma, kas izraisa ģenētisku traucējumu. Risks ir mazāks par 1% gadījumiem, kad pirmajam bērnam ir gēnu izdalīšanās vai viena vecāka parthenogenetiskā disosmija. Risks ir līdz 50%, ar nosacījumu, ka neveiksmi izraisa mutācija. Ar vecāku hromosomu pārvietošanu paliek risks līdz pat 25%.Jebkurā gadījumā vecākiem jāveic ģenētiskā pārbaude.
Prader-Willi sindroms un tā prognoze bieži saglabā runas aizkavēšanos, kā arī garīgo attīstību. Freim un Kerfs pētījumi parādīja, ka 5% pacientu ir vidēji izlūkošanas koeficients. Tas atbilst 85 vai vairāk punktiem IQ skalā.27% pacientu ir izlūkdatu līmenis par vidējo robežu, kas tiek lēsts 70-85 punkti.34% pacientu ir izlūkošanas līmenis, kas atbilst 50-70 punktiem.27% pacientu ir vidēji ilgstoši un novērtēti 35-70 punkti.5% pacientu ir spēcīga novecošanās un iegūst 20-35 punktus, un mazāk nekā 1% ir ievērojams kavēklis. Citi pētījumi liecina, ka 40% pacientu parāda vidējo vai samazināto izlūkošanas līmeni.
Bērniem ar Prader-Willi sindromu parasti ir laba ilglaicīga un vizuāla atmiņa. Viņi spēj iemācīties lasīt, ir pasīvi, bagāti vārdi, tomēr viņu runa bieži vien ir sliktāka nekā tās izpratne. Arī dzirdes atmiņa, rakstīšanas prasmes un matemātiskās prasmes, īslaicīga dzirdes un vizuālā atmiņa arī cieš un neatbilst vecuma standartiem.
Prader-Willi sindroms bieži vien ir saistīts ar palielinātu apetīti, jo pacientiem ir paaugstināts grilīna hormona līmenis asinīs. Pacientiem tipiska ir somatoliberīna zemā koncentrācija. Daži to asociē ar 15. hromosomu, kas saistīts ar hipotalāmu. Citi dati liecina par kopējā šūnu skaita samazināšanos, kā arī par oksitocīnu saturošām šūnām hipotalāmu paraventrikulārajos kodos. Bet interesanti, ir pierādījumi, kas liecina par pretējo: mirušo atklāšana ar šo slimību bieži nerada defektus hipotalāmā.