Amyloïdose: wat is het, oorzaken, symptomen, behandeling, prognose
Inhoud
- Wat is amyloïdose?
- Tekenen en symptomen
- Oorzaken en risicofactoren
- Getroffen populaties
- Verwante aandoeningen
- Diagnostiek
- Standaard behandelingen
- Voorspelling
Wat is amyloïdose?
Amyloïdose is een systemische ziekte die in vele soorten wordt onderverdeeld en wordt gekenmerkt door schade aan de parenchymale organen (d.w.z. de schildklier, longen, nieren, milt, lever). Het resultaat van onjuiste vorming en overmatige accumulatie in de intercellulaire ruimte van een complex, onoplosbaar eiwit met een laag molecuulgewicht, of zo eiwit-polysacharidecomplex genoemd, werkt als sclerose en weefselatrofie en leidt als gevolg daarvan tot een tekort aan bovenstaande organen.
Deze pathologie is relatief jong en werd geïdentificeerd door de Duitse wetenschapper M.Ya Schleiden. in 1983, wat de deelname van grove eiwitten aan de vorming van amyloïde aantoonde.
Soorten amyloïdose:
- AL-amyloïdose (primair) is het meest voorkomende type systemische amyloïdose. AL-amyloïdose is het gevolg van een afwijking (dyscrasie) van een type witte bloedcel, plasmacellen genaamd, in het beenmerg en is nauw geassocieerd met multipel myeloom.
- AA-amyloïdose (secundair) komt van het serum-inflammatoire eiwit amyloïde A. AA-amyloïdose komt voor in verband met een chronische ontstekingsziekte zoals reumatische aandoeningen, chronische inflammatoire darmziekte, tuberculose of empyeem.
- AF amyloïdose (mediterrane intermitterende koorts) — erfelijke vorm van amyloïdose, met een autosomaal recessief transmissiemechanisme. Dit type amyloïdose treft mensen die behoren tot bepaalde etnische groepen die langs de Middellandse Zeekust wonen (Sefardische joden, Grieken, Arabieren, Armeniërs). Er zijn variëteiten van amyloïdose die kenmerkend zijn voor een bepaald geografisch gebied: "Portugese amyloïdose" (met een overheersende laesie van de zenuwen van de onderste ledematen), "Amerikaanse amyloïdose "(met een overheersende laesie van de zenuwen van de bovenste ledematen), familiale nefropathische amyloïdose of" Engelse amyloïdose ", optredend met symptomen van urticaria, doofheid en koorts.
- AH amyloïdose - uitsluitend waargenomen bij patiënten die hemodialyse ondergaan. De pathogenese houdt verband met het feit dat microglobuline bèta-2 klasse MHC I, normaal gebruikt door de nieren, niet wordt gefilterd in de hemodialysator en zich ophoopt in het lichaam.
- AE amyloïdose - een vorm van lokale amyloïdose die zich bij sommige tumoren ontwikkelt, bijvoorbeeld bij medullair carcinoom van C-cellen van de schildklier. In dit geval zijn pathologische fragmenten van calcitonine de voorloper van amyloïde.
- ASC1 amyloïdose - seniele systemische amyloïdose. De voorloper van het fibrillaire eiwit ASC1 is serum prealbumine. Er wordt aangenomen dat in verband met de schending van het metabolisme van prealbumine op oudere en seniele leeftijd, de neiging om een mutant eiwit te vormen uit het circulerende bloed toeneemt.
- Aβ-amyloïdose - waargenomen bij ziekte van Alzheimer, soms familiezaken.
- AIAPP-amyloïdose - waargenomen bij type 2 diabetes en insulineoom.
- Finse type amyloïdose - een zeldzaam type ziekte dat optreedt als gevolg van een mutatie in het GSN-gen, dat codeert voor het jelsoline-eiwit.
Lees ook:Vasculitis (angiitis): wat is het, oorzaken, symptomen (foto), soorten angiitis, behandeling
Tekenen en symptomen
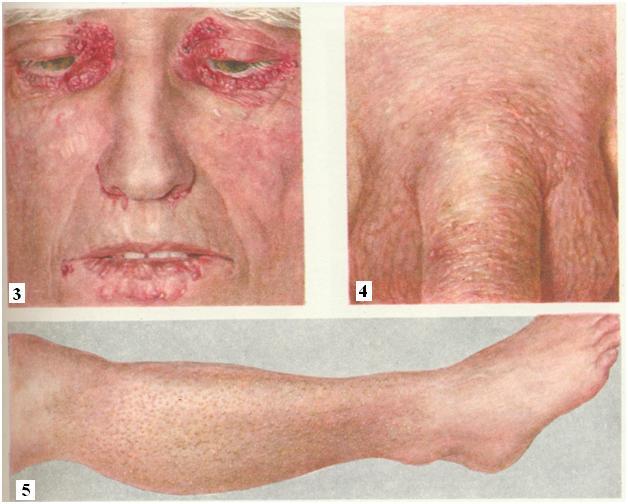
De klinische symptomen van amyloïdose kunnen variëren en zijn afhankelijk van de ernst en lokalisatie. amyloïde afzettingen, biochemische samenstelling van amyloïde, duur van de ziekte, mate van disfunctie organen. De latente periode van amyloïdose, wanneer glycoproteïne-afzettingen alleen microscopisch kunnen worden gedetecteerd, verschilt niet in de ontwikkeling van significante tekenen. Naarmate het functionele falen van het aangetaste orgaan vordert, nemen de klinische symptomen van de ziekte toe.
Met amyloïdose van de nieren het huidige stadium van matige proteïnurie op lange termijn wordt vervangen door het optreden van nefrotisch syndroom. De overgang naar het uitgebreide stadium kan gepaard gaan met een uitgestelde bijkomende infectie, vaccinatie, hypothermie, verergering van de onderliggende ziekte. De patiënt heeft een geleidelijke toename van oedeem, het optreden van nefrogene arteriële hypertensie en nierfalen. Soms ontwikkelt zich nierveneuze trombose. Massaal eiwitverlies gaat gepaard met de ontwikkeling van hypoproteïnemie, hyperfibrinogenemie, hyperlipidemie, azotemie. Micro-, soms grove hematurie, leukocyturie worden gevonden in de urine.
Met amyloïdose van het hart ontwikkeling van restrictieve cardiomyopathie met typische klinische symptomen - cardiomegalie wordt opgemerkt, aritmievordert hartfalen. De patiënt heeft kortademigheid, zwelling, zwakte die zelfs bij lichte lichamelijke inspanning optreedt. In zeldzame gevallen, met amyloïdose van het hart, treedt polyserositis op, gemanifesteerd door het optreden van ascites, exsudatieve pleuritis en pericarditis.

Schade aan het spijsverteringsstelsel met amyloïdose wordt het gekenmerkt door amyloïde infiltratie van de tong, slokdarm, maag, darmen. Gastro-intestinale bloedingen zijn mogelijk. Met amyloïde infiltratie van de lever, hepatomegalie, cholestase, portale hypertensie. De nederlaag van de pancreas in deze pathologie kan worden vermomd als: chronische pancreatitis.
Huid amyloïdose gekenmerkt door het verschijnen van meerdere wasachtige plaques in het gezicht, nek, natuurlijke huidplooien. Uiterlijk kunnen huidlaesies lijken op: sclerodermie, neurodermitis of lichen planus.

Bij gewrichtsschade is de ontwikkeling van symmetrische polyartritis, carpaaltunnelsyndroom, humerus-scapulaire periartritis en myopathie typisch. Bepaalde vormen van amyloïdose die gepaard gaan met schade aan het zenuwstelsel kunnen gepaard gaan met de ontwikkeling polyneuropathie, verlamming van de onderste ledematen, hoofdpijn, duizeligheid, orthostatische hypotensie, zweten, Dementie.
Lees ook:Artrose (artrose)
Oorzaken en risicofactoren
De redenen voor de ontwikkeling van primaire amyloïdose worden momenteel niet volledig begrepen. Het bleek dat de ontwikkeling van secundaire amyloïdose meestal gepaard gaat met chronische infectieziekten (tuberculose, syfilis, actinomycose) en pyo-inflammatoire ziekten (osteomyelitis, bronchiëctasie, bacterieel endocarditis), minder vaak wordt de ziekte geassocieerd met tumorprocessen (lymfogranulomatose, leukemie, kanker van de viscerale organen).
De ontwikkeling van een reactieve vorm van amyloïdose treft personen die lijden aan atherosclerose, psoriasis, reumatische aandoeningen, zoals reumatoïde artritis, spondylitis ankylopoeticachronische ontstekingsziekten zoals niet-specifieke colitis ulcerosa, ziekte van Crohn, met multisysteemlaesies zoals Ziekte van Whipple of sarcoïdose.
Risicofactoren voor amyloïdose kunnen hyperglobulinemie, verminderde werking van cellulaire immuniteit, erfelijke aanleg zijn
Getroffen populaties
Naar schatting worden jaarlijks ongeveer 4.000 nieuwe gevallen van AL-amyloïdose gemeld, hoewel de werkelijke incidentie iets hoger kan zijn als gevolg van onvoldoende diagnose. Hoewel wordt aangenomen dat de incidentie bij mannen en vrouwen hetzelfde is, is ongeveer 60% van de in de centra opgenomen patiënten mannen. AL-amyloïdose komt voor bij mensen van in de twintig, maar wordt meestal gediagnosticeerd tussen de leeftijd van 50 en 65 jaar.
Mensen met een risico op het ontwikkelen van AA-amyloïdose zijn onder meer mensen met chronische ontstekingsaandoeningen zoals: Reumatoïde artritis, psoriatische arthritis, chronische juveniele artritis, spondylitis ankylopoetica bij kinderen, inflammatoire darmziekte.
Mensen met chronische infectieziekten zoals: tuberculose, lepra, bronchiëctasie, chronische osteomyelitis en chronische pyelonefritislopen ook gevaar. Secundaire amyloïdose (AA) komt voor bij minder dan 5% van de mensen met deze aandoeningen.
Verwante aandoeningen
De volgende aandoeningen kunnen in verband worden gebracht met amyloïdose. Amyloïdose kan optreden in combinatie met of als gevolg van de volgende aandoeningen:
multipel myeloom, lymfoom, Hodgkin-lymfoom, medullair schildkliercarcinoom, ziekte van Whipple, ziekte van Crohn, osteomyelitis, reumatoïde artritis, spondylitis ankylopoetica, syndroom van Reiter, artritis psoriatica, tuberculose, macrobolisme, congenitale purulent-veneuze ziekte (congenitale intestinale hyperplasie, erfelijke congenitale rigidrocytogenese)
Lees ook:Temporale arteritis
Diagnostiek
Vooral in het geval van AL-amyloïdose is een vroege diagnose de sleutel tot overleving en herstel van de kwaliteit van leven na behandeling. De diagnose amyloïdose wordt vermoed na gedetailleerde anamnese en klinische evaluatie, maar vereist abdominale vetaspiratie en/of biopsie van het betrokken orgaan.
Als de ziekte klinisch wordt vermoed, geeft een biopsie van het aangetaste orgaan het beste resultaat.
Het biopsiemateriaal wordt onder een microscoop onderzocht en gekleurd met een kleurstof die groen afgeeft wanneer bekeken onder een polariserende microscoop als amyloïde aanwezig is. Wanneer amyloïdose wordt gediagnosticeerd met een weefselbiopsie, is het belangrijk dat het slachtoffer nader wordt onderzocht om te bepalen welke organen zijn aangetast.
Bij mensen die langdurig dialyse ondergaan of in het eindstadium nierfalenlaboratoriumtests kunnen worden gedaan die bloed- of urinemonsters kunnen analyseren om verhoogde niveaus van B2M-eiwit te detecteren.
Standaard behandelingen
In de meeste gevallen wordt amyloïdose thuis behandeld. In aanwezigheid van complicaties kan de patiënt worden opgenomen in het ziekenhuis.
Therapie voor amyloïdose omvat het nemen van medicijnen en het volgen van een aantal aanbevelingen van de arts. Maar in ernstige gevallen wordt de milt verwijderd en kan een nier- of levertransplantatie nodig zijn.
De lijst met medicijnen hangt af van de lokalisatie van afzettingen, de mate van schade aan het lichaam, bestaande complicaties. Bij secundaire amyloïdose is dus een specifieke behandeling van de primaire ziekte noodzakelijk. Bovendien worden medicijnen voorgeschreven om symptomen te elimineren.
Ook krijgt de patiënt vaak een speciaal dieet te zien (beperking van de inname van eiwit en zout).
Er is geen specifiek profylactisch programma voor amyloïdose, omdat de exacte oorzaken van de ziekte onbekend zijn.
Voorspelling
De prognose hangt af van het type amyloïdose en het aangetaste orgaansysteem, maar met de juiste pathogenetische behandeling en ondersteunende therapie is de levensverwachting van veel patiënten vrij lang.
De gemiddelde levensverwachting van patiënten met AA-amyloïdose is vermoedelijk 10 jaar. De meest voorkomende doodsoorzaak is nierfalen. Onbehandelde patiënten met AL-amyloïdose leven ongeveer een jaar vanaf de diagnose. De prognose verergert de schade aan het cardiovasculaire systeem.