Lysosomale lagringssykdommer: hva er det, symptomer, behandling, prognose
Innhold
- Hva er lysosomale lagringssykdommer?
- Hva er lipider?
- Hvordan arves lysosomale lagringssykdommer?
- Typer og symptomer på lysosomale lagringssykdommer
Hva er lysosomale lagringssykdommer?
Lysosomale lagringssykdommer, er en gruppe arvelige metabolske sykdommer der skadelige mengder fettstoffer (lipider) akkumuleres i forskjellige celler og vev i kroppen.
Personer med disse lidelsene får enten ikke nok av et av enzymene som trengs for å bryte ned (metabolisere) lipider, eller de produserer enzymer som ikke fungerer som de skal.
Over tid kan denne overdrevne akkumuleringen av fett føre til permanent skade på celler og vev, spesielt i hjernen, det perifere nervesystemet (nerver fra ryggmargen til resten av kropp), lever, milt og beinmarg.
Hva er lipider?

Lipider er fettlignende stoffer som er viktige deler av membranene inne i og mellom cellene og i myelinkappen, som dekker og beskytter nervene. Lipider inkluderer oljer, fettsyrer, voks, steroider (som kolesterol og østrogen) og andre beslektede forbindelser.
Disse fete stoffene lagres naturlig i cellene, organene og vevene i kroppen. Små kropper i celler som kalles lysosomer, omdanner eller metaboliserer jevnlig lipider og proteiner til mindre komponenter for å gi kroppen energi. Lidelser der intracellulært materiale, som ikke kan metaboliseres, forblir i lysosomer, kalles lysosomale lagringssykdommer.
I tillegg til sykdommer forbundet med lipidakkumulering, inkluderer andre lysosomale sykdommer forbundet med akkumulering mukolipidoser, der celler og vev akkumuleres en overdreven mengde lipider med sukkermolekyler festet til dem, så vel som mukopolysakkaridoser, der en overdreven mengde stort, komplekst sukker akkumuleres molekyler.
Hvordan arves lysosomale lagringssykdommer?

Lysosomale lagringssykdommer arves fra en eller begge foreldrene som bærer et defekt gen som regulerer et spesifikt lipidmetaboliserende enzym i en klasse celler i kroppen. De kan arves på to måter:
- Autosomal recessiv arv oppstår når begge foreldrene bærer og videreformidler en kopi av det defekte genet, men ingen av foreldrene påvirkes av lidelsen. Hvert barn født av disse foreldrene har 25 prosent sjanse for å arve begge kopiene av det defekte genet, 50 prosent sjanse for å være en forelderlignende transportør og 25 prosent sjanse for ikke å arve noen kopi av den defekte genet. Barn av begge kjønn kan arves på en autosomal recessiv måte.
- X-koblet (eller kjønnsrelatert) recessiv arv oppstår når mor bærer det berørte genet på X -kromosomet. X- og Y -kromosomene er involvert i kjønnsbestemmelse. Kvinner har to X -kromosomer, mens menn har ett X -kromosom og ett Y -kromosom. Sønner av kvinnelige bærere har 50 prosent sjanse for å arve og påvirke lidelsen, ettersom sønner mottar ett X -kromosom fra moren og ett Y -kromosom fra faren. Døtre har 50 prosent sjanse for å arve det berørte X -kromosomet fra moren og er bærere eller lett påvirket. Berørte menn overfører ikke sykdommen til sønnene, men døtrene deres vil være bærere og bærere av sykdommen.
Typer og symptomer på lysosomale lagringssykdommer
Gauchers sykdom - den vanligste av de lysosomale lagringssykdommene. Sykdommen er forårsaket av mangel på enzymet glukocerebrosidase, noe som fører til akkumulering av glukoserebroside i mange vev. Fett materiale kan samle seg i hjernen, milten, leveren, nyrene, lungene og benmargen.
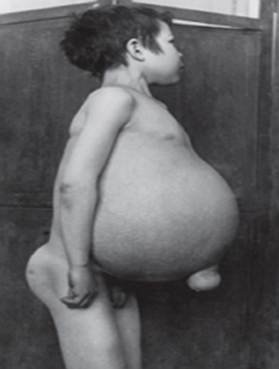
Symptomer kan omfatte hjerneskade, utvidelse av milten lever, nedsatt leverfunksjon, skjelettplager og beinskader som kan forårsake smerter og brudd, hevelse i lymfeknuter og (noen ganger) tilstøtende ledd, oppblåsthet, brunaktig hudtone, anemi, lave blodplater og gule flekker på øynene.
De som er hardest rammet kan også være mer utsatt for infeksjoner. Sykdommen rammer menn og kvinner likt.
Gauchers sykdom har tre generelle kliniske undertyper:
- Type 1 (Eller ikke nevronopatisk type) er den vanligste sykdomsformen i USA og Europa. Hjernen påvirkes ikke, men det kan være skade på lungene og i sjeldne tilfeller nyrene. Symptomer kan begynne tidlig i livet eller i voksen alder og inkluderer en forstørret lever og alvorlig forstørrelse av milten, noe som kan føre til brudd og ytterligere komplikasjoner. Skjelettlidelse og bein sykdom kan være omfattende. Folk i denne gruppen blåser vanligvis lett på grunn av lavt antall blodplater i blodet. De kan også oppleve tretthet på grunn av anemi. Avhengig av sykdomsutbruddet og alvorlighetsgraden, mennesker med type 1 kan leve til voksen alder. Mange pasienter har mild sykdom eller viser ikke symptomer. Selv om 1type Gauchers sykdom er vanlig blant mennesker med Ashkenazi -jødisk arv og kan påvirke mennesker med alle etniske bakgrunner.
- Type 2 (eller akutt nevropatisk barndom Gauchers sykdom) begynner vanligvis innen 3 måneder etter fødselen. Symptomene inkluderer omfattende og progressiv hjerneskade, spastisitet, anfall, stive lemmer, forstørret lever (lever hepatomegali) og milt (splenomegali), unormal øyebevegelse og dårlig evne til å suge og svelge. Berørte barn dør vanligvis før 2 år.
- Type 3 (kronisk nevropen form) kan begynne når som helst i barndommen eller til og med i voksen alder. Det er preget av sakte progressive, men mindre alvorlige nevrologiske symptomer sammenlignet med akutt Gauchers sykdom 2 typer. Store symptomer inkluderer øyebevegelsesforstyrrelser, kognitive underskudd, dårlig koordinasjon, anfall, utvidelse av milten og / eller leveren, skjelettlidelser, blodforstyrrelser inkludert anemi og problemer med puster. Nesten alle med Gauchers sykdom 3 typer, som får enzymbyttebehandling vil nå voksenlivet.
Les også:Meslinger hos barn, foto fra startfasen
For pasienter 1. og 3. type Enzymerstatningsterapi, gitt intravenøst annenhver uke, kan dramatisk redusere størrelsen på leveren og milten, redusere avvik i skjelettet og reversere andre manifestasjoner. En vellykket benmargstransplantasjon behandler de nevrologiske manifestasjonene av sykdommen. Imidlertid medfører denne prosedyren betydelige risikoer og gjøres sjelden hos personer med Gauchers sykdom.
I sjeldne tilfeller kan det være nødvendig med kirurgi for å fjerne hele eller deler av milt (hvis en person har et svært lavt antall blodplater eller når et forstørret organ i stor grad påvirker en persons komfort). Blodoverføring kan være til nytte for noen med anemi. Andre kan trenge leddskifteoperasjon for å forbedre mobiliteten og livskvaliteten. Det er foreløpig ingen effektiv behandling for hjerneskade som kan oppstå hos personer med type 2 og type 3 Gauchers sykdom.
Niemann-Pick sykdom er en gruppe autosomale recessive lidelser forårsaket av akkumulering av fett og kolesterol i leverceller, milt, benmarg, lunger og i noen tilfeller hjernen.
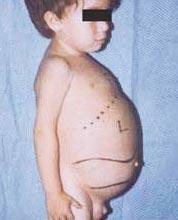
Nevrologiske komplikasjoner kan inkludere ataksi (mangel på muskelkoordinering som kan påvirke gåing, skriving og spising, blant andre funksjoner), øyelammelse, hjernedegenerasjon, læringsproblemer, spastisitet, problemer med å mate og svelge, sløret tale, tap av muskeltonus, overfølsomhet for berøring og noe uklarhet i hornhinnen på grunn av overdreven akkumulering materialer. Hos 50% av de berørte menneskene utvikler det seg en karakteristisk kirsebærrød glorie rundt midten av netthinnen, som kan sees av en lege med et spesielt instrument.
Niemann-Pick sykdom er klassifisert i tre generelle undertyper:
- Type A, den alvorligste formen, begynner tidlig i barndommen. Babyer virker normale ved fødselen, men etter 6 måneder utvikler dyp hjerneskade, forstørret lever og milt og hovne lymfeknuter og noder under huden (xanthomas). Milten kan forstørre 10 ganger sin normale størrelse og kan sprekke og forårsake blødning. Disse barna blir stadig svakere, mister motorisk funksjon, kan bli anemiske og utsatt for tilbakevendende infeksjoner. De lever sjelden mer enn 18 måneder. Denne sykdomsformen er mest vanlig i jødiske familier.
- Type B(eller ungdomsdebut) påvirker vanligvis ikke hjernen, men de fleste barn utvikler ataksi, skade på nervene som forlater ryggmargen (perifer nevropati), og lungevansker som utvikler seg med alderen. Forstørrelse av lever og milt er typisk for ungdom. Folk med type B kan leve relativt lenge, men mange krever livslang oksygenbehandling på grunn av lungeskade. Niemann-Pick type A og B er et resultat av en opphopning av et fettstoff som kalles sfingomyelin på grunn av mangel på et enzym som kalles sphingomyelinase.
- Type C kan dukke opp tidlig i livet eller utvikle seg i ungdomsårene eller til og med i voksen alder. Niemann-Pick sykdom type C er ikke forårsaket av sphlingomyelinase -mangel, men av mangel på NPC1- eller NPC2 -proteiner. Som et resultat akkumuleres forskjellige lipider og spesielt kolesterol i nerveceller og forårsaker funksjonsfeil. Hjernens involvering kan være omfattende, noe som resulterer i manglende evne til å se opp og ned, problemer med å gå og svelge, progressivt hørselstap og progressiv demens. Folk med type C har bare mild forstørrelse av milten og leveren. Forventet levetid for mennesker med type C varierer betraktelig. Noen mennesker dør i barndommen, mens andre som er mindre påvirket også kan leve i voksen alder.
Det er foreløpig ingen kur mot Niemann-Pick sykdom. Behandlingen er støttende. Barn dør vanligvis av infeksjon eller progressivt nevrologisk tap. Det har vært tilfeller av benmargstransplantasjon hos flere pasienter med type B med blandede resultater.
Fabrys sykdom også kjent som alfa-galaktosidase-A-mangel, forårsaker opphopning av fettstoffer i det autonome nervesystemet (den delen av nervesystemet som kontrollerer ufrivillige funksjoner som pust og hjerteslag), i øyne, nyrer og kardiovaskulær system.
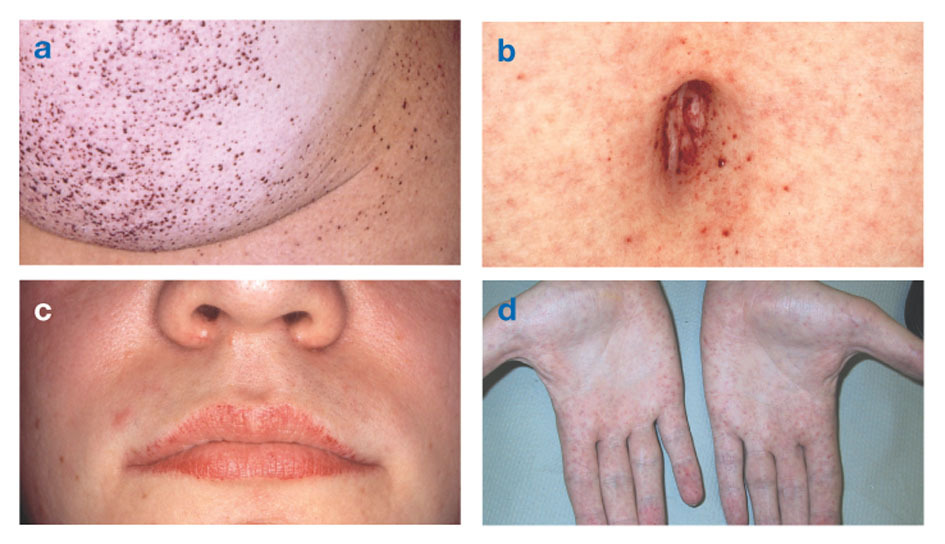
Fabrys sykdom er den eneste X-koblede lipidsykdommen. Sykdommen rammer hovedsakelig menn, selv om en mildere og mer variabel form forekommer hos kvinner. Sjelden har berørte kvinner alvorlige symptomer som ligner på de som er sett hos menn med lidelsen. Symptomutbruddet oppstår vanligvis i barndommen eller ungdomsårene.
Nevrologiske tegn inkluderer brennende smerter i armer og ben som blir verre i varmt vær eller etterpå trening, samt akkumulering av overflødig materiale i de gjennomsiktige lagene på hornhinnen (noe som fører til grums, men uten endringer i synet). Fettakkumulering i blodkarets vegger kan forstyrre sirkulasjonen og sette en person i fare slag eller hjerteinfarkt. Andre symptomer inkluderer et forstørret hjerte, progressivt nyresviktsom fører til nyresykdom i sluttstadiet, gastrointestinale problemer, redusert svette og feber.
Les også:Rakitt hos spedbarn: årsaker, symptomer og behandling
Angiokeratomer (små, ikke-kreft, rødlig-lilla hevede flekker på huden) kan utvikle seg i nedre torso og bli mer vanlig med alderen.
Personer med Fabrys sykdom dør ofte for tidlig av komplikasjoner fra hjertefeil, nyresvikt eller slag. Medisiner som fenytoin og karbamazepin er ofte foreskrevet for å behandle smerten som følger med Fabrys sykdom, men ikke for å kurere den. Metoklopramid eller Lipisorb (kosttilskudd) kan lindre gastrointestinal forstyrrelse forekommer ofte hos personer med Fabry sykdom, og noen mennesker kan trenge en nyretransplantasjon eller dialyse. Enzymbytte kan redusere lagring, lindre smerter og bevare organfunksjonen hos noen mennesker med Fabrys sykdom.
Farbers sykdom også kjent som Farbers lipogranulomatose, beskriver en gruppe sjeldne autosomale recessive lidelser som forårsaker akkumulering av fettstoffer i ledd, vev og sentralnervesystemet. Det påvirker både menn og kvinner. Sykdommen begynner vanligvis tidlig i barndommen, men kan oppstå senere i livet.
Barn med klassisk Farbers sykdom utvikler seg nevrologiske symptomer, som kan omfatte økt søvnighet og slapphet, og problemer med svelging. Lever, hjerte og nyrer kan også påvirkes. Andre symptomer kan omfatte leddkontrakturer (kronisk forkortelse av muskler eller sener rundt leddene), oppkast, leddgikt, forstørrelse lymfeknuter, leddbetennelse, heshet og klumper under huden som tykner rundt leddene etter hvert som det utvikler seg sykdommer.
Berørte personer med pustevansker kan trenge å sette inn et pusteslange. De fleste barn med denne tilstanden dør etter 2 år, vanligvis fra lungesykdommer. I en av de alvorligste sykdomsformene kan en forstørret lever og milt diagnostiseres kort tid etter fødselen. Babyer født med denne sykdomsformen dør vanligvis innen 6 måneder.
Farbers sykdom skyldes mangel på et enzym som kalles ceramidase. Det er foreløpig ingen spesifikk behandling for Farbers sykdom. Kortikosteroider kan foreskrives for å lindre smerter. Benmargstransplantasjon kan redusere granulomer (små masser av betent vev) hos mennesker med mild betennelse eller hos pasienter uten komplikasjoner i lunge- eller nervesystemet. Hos eldre mennesker kan granulomer krympe eller fjernes kirurgisk.
Gangliosidose består av to forskjellige grupper av genetiske sykdommer. Begge er autosomale recessive og påvirker menn og kvinner likt.
GM1 gangliosidose.
GM1 gangliosidose er forårsaket av mangel på enzymet beta-galaktosidase, noe som resulterer i en unormal akkumulering av sure lipidmaterialer, spesielt i nerveceller i sentral- og perifer nerve systemer. GM1 gangliosidose har tre kliniske manifestasjoner:
- GM1 (den alvorligste undertypen, som forekommer like etter fødselen) kan omfatte nevrodegenerasjon, anfall, forstørret lever og milt, grovhet i ansiktstrekk, uregelmessigheter i skjelettet, leddstivhet, oppblåsthet, muskelsvakhet, overdreven skremmende respons og problemer med gangart. Omtrent halvparten av de berørte utvikler kirsebærrøde flekker i øyet. Barn kan være døve og blinde etter 1 år og dør ofte innen 3 år av hjertekomplikasjoner eller lungebetennelse.
- Sent barn GM1 gangliosidose begynner vanligvis mellom 1 og 3 år. Nevrologiske symptomer inkluderer ataksi, anfall, demens og vanskeligheter med å snakke.
- GM1 gangliosidose utvikler seg mellom 3 og 30 år. Symptomer inkluderer redusert muskelmasse (muskelatrofi), nevrologiske komplikasjoner som er mindre alvorlige og utvikler seg saktere enn andre former lidelser, uklarhet i hornhinnen hos noen mennesker og vedvarende muskelsammentrekninger som forårsaker vridning og repeterende bevegelser eller unormale stillinger (dystoni). Angiokeratomer kan utvikle seg i den nedre stammen av kroppen. Størrelsen på leveren og milten er normal hos de fleste berørte mennesker.
GM2 gangliosidose.
GM2 gangliosidose får også kroppen til å akkumulere overflødige sure fettsyrer i vev og celler, først og fremst nerveceller. Disse lidelsene skyldes mangel på enzymet beta-heksosaminidase. GM2 lidelser inkluderer:
- Tay-Sachs sykdom (også kjent som GM2 gangliosidose variant B) og dens variantformer er forårsaket av mangel på enzymet heksosaminidase A. Berørte barn utvikler seg normalt i løpet av de første månedene av livet. Symptomer begynner ved 6 måneders alder og inkluderer gradvis tap av mental kapasitet, demens, redusert øyekontakt, økt et skremmende svar på støy, progressivt hørselstap som fører til døvhet, problemer med å svelge, blindhet, kirsebærrøde flekker på netthinnen, og noen lammelse. Beslag kan begynne i det andre året av et barns liv. Babyer kan til slutt bli avhengige av fôringsrøret, og dør ofte i en alder av 4 år av tilbakevendende infeksjoner. Dessverre er det ingen spesiell behandling. Antikonvulsiva kan i utgangspunktet kontrollere anfall. Andre støttende behandlinger inkluderer riktig ernæring og hydrering, og metoder for å holde luftveiene åpne. En sjelden form for lidelsen som kalles sent oppstått Tay-Sachs sykdom, forekommer hos mennesker mellom 20 og 30 år og er preget av en ustabil gang og progressiv nevrologisk forverring.
- Sandhoffs sykdom (Alternativ AB) er en alvorlig form for Tay-Sachs sykdom. Begynnelsen oppstår vanligvis ved 6 måneders alder og er ikke begrenset til noen etnisk gruppe. Nevrologiske tegn kan omfatte progressiv forverring av sentralnervesystemet, motor svakhet, tidlig blindhet, alvorlig skremt reaksjon på lyd, spasticitet, sjokk eller rykninger muskler (myoklonus), epilepsi, et unormalt forstørret hode (makrocefali) og røde flekker i øyet. Andre symptomer kan omfatte hyppige luftveisinfeksjoner, hjerteklump, dukkeaktige ansiktstrekk og forstørrelse av lever og milt. Det er ingen spesifikk behandling for Sandhoff sykdom. Som med Tay-Sachs sykdom inkluderer støttebehandling å holde luftveiene åpne hele tiden, samt riktig ernæring og hydrering. Antikonvulsiva kan i utgangspunktet kontrollere epilepsi (anfall).
Les også:Fostervannemboli
Krabbes sykdom (også kjent som globulær celle leukodystrofi og galaktosylseramid lipidose) er en autosomal recessiv sykdom forårsaket av mangel på enzymet galaktocerebrosidase. Sykdommen rammer oftest spedbarn som begynner før 6 måneder, men kan oppstå i ungdomsårene eller i voksen alder.
Akkumulering av ufordøyd fett påvirker veksten av nervens beskyttende kappe (myelinskede) og forårsaker alvorlig svekkelse av mentale og motoriske ferdigheter. Andre symptomer inkluderer muskelsvakhet, redusert evne til å strekke muskler (hypotoni), muskelspenninger (spastisitet), plutselig sjokk eller rykninger i lemmer (myokloniske anfall), irritabilitet, uforklarlig feber, døvhet, blindhet, lammelse og problemer med å svelge. Langsiktig vekttap kan også forekomme.
Sykdommen kan diagnostiseres ved enzymtesting og identifisering av dens karakteristiske cellegruppering inn i globoidlegemer i hjernens hvite substans, demyelinisering av nerver og degenerasjon og ødeleggelse av hjerneceller. Hos spedbarn er sykdommen vanligvis dødelig før 2 år. Personer med en mer avansert sykdomsform har et mildere sykdomsforløp og lever betydelig lenger.
Det er ikke utviklet noen spesifikk behandling for Krabbes sykdom, selv om tidlig benmargstransplantasjon kan hjelpe noen mennesker. Personer med en mer avansert sykdomsform har et mildere sykdomsforløp og lever betydelig lenger.
Metakromatisk leukodystrofi, eller MLD, er en gruppe lidelser preget av opphopning av fettstoffer i det hvite stoffet i sentralnervesystemet og i perifere nerver og til en viss grad i nyrene. I likhet med Krabbes sykdom påvirker MLD myelin, som dekker og beskytter nervene. Denne autosomale recessive lidelsen skyldes mangel på enzymet arylsulfatase A. Denne lidelsen rammer både menn og kvinner.
Metakromatisk leukodystrofi har tre karakteristiske former: sent spedbarn, ung og voksen.
- Sent MLD for spedbarn starter vanligvis mellom 12 og 20 måneder etter fødselen. Barn kan virke normale i begynnelsen, men har problemer med å gå og har en tendens til å falle, ledsaget av periodiske smerter i armer og ben. progressivt synstap som fører til blindhet, forsinkelser i utviklingen og tap av tidligere ervervede milepæler, nedsatt svelging, anfall og demens opptil 2 år gammel. Barn utvikler også progressiv muskelsvinn og svakhet og mister til slutt evnen til å gå. De fleste barn med denne formen for lidelsen dør innen 5 år.
- Juvenil MLD begynner vanligvis mellom 3 og 10 år. Symptomer inkluderer nedsatt skoleprestasjon, psykisk svekkelse, ataksi, anfall og demens. Symptomene utvikler seg, og døden skjer 10–20 år etter sykdomsutbruddet.
- Voksenform. Symptomer voksne oppstår etter 16 år og kan omfatte ataksi, anfall, unormale skjelvinger i lemmer (skjelvinger), nedsatt konsentrasjon, depresjon, psykiske lidelser og demens. Døden skjer vanligvis 6–14 år etter symptomdebut.
MLD kan ikke kureres. Behandlingen er symptomatisk og støttende. Benmargstransplantasjon kan i noen tilfeller forsinke utviklingen av sykdommen. Det er gjort betydelige fremskritt med genterapi i dyremodeller med MLD og i kliniske studier.
Wolmans sykdomogså kjent som lysosomal syre lipase mangeler en alvorlig lipidlagringsforstyrrelse som vanligvis er dødelig ved 1 års alder. Denne autosomale recessive lidelsen er preget av en opphopning av kolesterolestere (vanligvis transportformen av kolesterol) og triglyserider (den kjemiske formen der fett finnes i kroppen), som kan akkumuleres betydelig og forårsake celleskader og tekstiler.
Både menn og kvinner lider av denne lidelsen. Spedbarn er normale og aktive ved fødselen, men utvikler raskt progressiv psykisk svekkelse, en forstørret lever og sterkt forstørret milt, oppblåsthet, gastrointestinale problemer. gulsott, anemi, oppkast og kalsiumavleiringer i binyrenefår dem til å stivne.
En annen type lysosomal syre lipase mangel er kolesterolester lagringssykdom. Denne ekstremt sjeldne sykdommen oppstår som et resultat av lagring av kolesterolestere og triglyserider i blodceller, lymfe og lymfoide vev. Hos barn i voksen alder forstørres leveren, noe som fører til skrumplever og kronisk leversvikt. Barn kan også ha kalsiumavsetninger i binyrene, og gulsott kan utvikle seg i de senere stadiene av sykdommen.
For tiden studeres spørsmålet om enzymbytte aktivt i Wolmans sykdom og i akkumulering av en kolesterolester.