Síndrome de Waardenburg: o que é, sintomas, tratamento, prognóstico
Contente
- O que é a síndrome de Waardenburg?
- sinais e sintomas
- Causas
- Tipo de herança
- Populações afetadas
- Distúrbios sintomáticos
- Diagnóstico
- Tratamentos padrão
- Previsão
O que é a síndrome de Waardenburg?
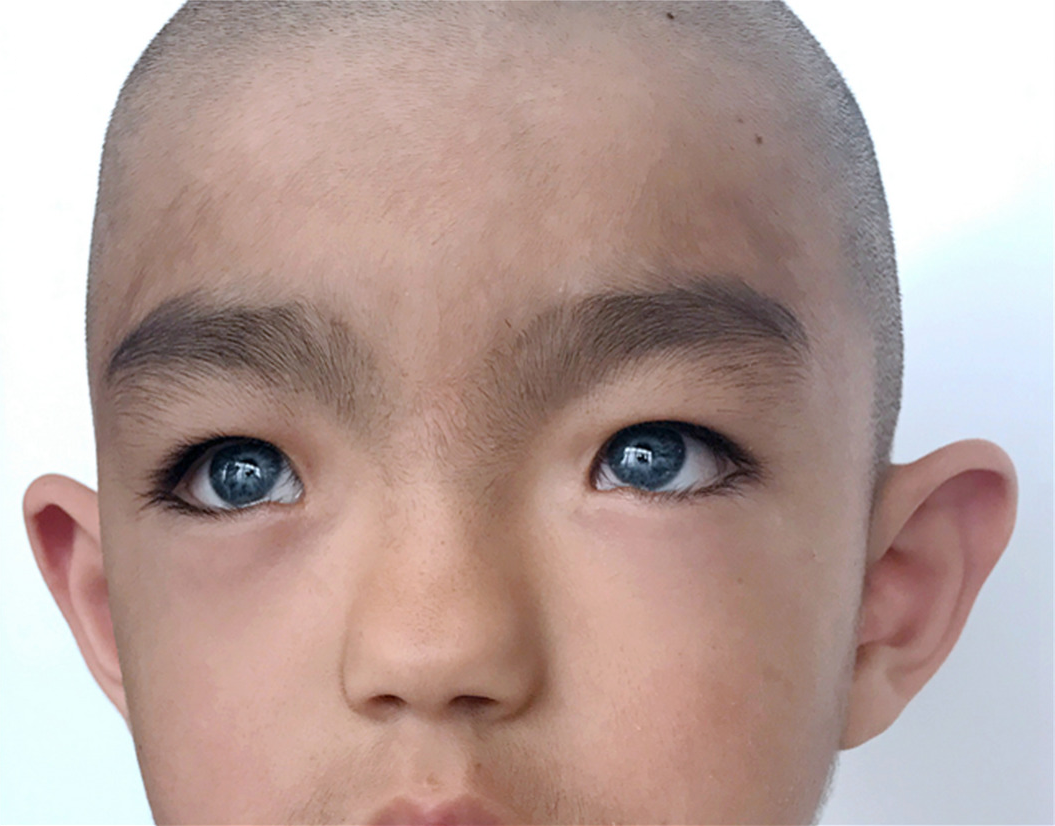
Síndrome de Waardenburg É uma doença genética que pode se manifestar no nascimento (congênita). A gama e a gravidade dos sintomas e sinais associados podem variar muito de caso para caso. No entanto, os sinais primários geralmente incluem anormalidades faciais características; coloração (pigmentação) anormalmente reduzida do cabelo, pele e / ou íris de ambos os olhos; e / ou surdez congênita. Mais especificamente, alguns pacientes podem ter uma ponte do nariz invulgarmente larga devido ao deslocamento lateral (lateral) dos cantos internos dos olhos (telecante). Além disso, as anormalidades de pigmentação podem incluir uma mecha branca de cabelo crescendo sobre a testa; envelhecimento prematuro ou branqueamento do cabelo; diferenças na cor de duas íris ou em partes diferentes da mesma íris (heterocromia da íris); e / ou áreas irregulares e anormalmente claras (despigmentadas) da pele (leucodermia). Algumas pessoas também podem ter deficiência auditiva devido a anormalidades no ouvido interno (perda auditiva neurossensorial).
Os pesquisadores descreveram vários tipos de síndrome de Waardenburg (doravante abbr. SV) com base em sintomas associados e dados genéticos específicos. Por exemplo, a síndrome de Waardenburg tipo I (CB1) está geralmente associada ao deslocamento lateral do canto interno do olho (isto é, telescópico), mas o tipo II (CB2) não está associado a esta característica. Além disso, CB1 e CB2 são conhecidos por serem causados por alterações (mutações) em diferentes genes. Outra forma, conhecida como tipo III (CB3), em que características faciais, olhos (ocular) e distúrbios auditivos podem estar associados a malformações características dos braços e das mãos (extremidades superiores). Uma quarta forma, conhecida como CB4 ou doença de Waardenburg-Hirschsprung, pode ter sinais primários de SV em combinação com Doença de Hirschsprung. Este último é um distúrbio digestivo (gastrointestinal) em que não há grupos de corpos especializados de células nervosas na parede muscular lisa (involuntária) da espessura intestinos.
Na maioria dos casos, a síndrome de Waardenburg é transmitida de maneira autossômica dominante. Vários genes de doenças diferentes foram identificados e podem causar a síndrome de Waardenburg em certos indivíduos ou famílias (parentes).
sinais e sintomas
Os principais sinais da síndrome de Waardenburg podem incluir:
- anomalias faciais características (ver. Foto);
- distúrbios de pigmentação (hipopigmentação) de cabelo, pele e / ou íris ou íris de ambos os olhos (parcial albinismo);
- perda auditiva congênita.
No entanto, como mencionado anteriormente, os sintomas e sinais associados podem ser muito diferentes, inclusive entre os membros da família afetados. Por exemplo, enquanto alguns pacientes podem ter apenas uma característica, outros podem ter várias anormalidades relacionadas à doença.
Em alguns pacientes com síndrome de Waardenburg (VS), ocorre um deslocamento anormal dos cantos internos laterais (laterais) dos olhos, formado pela junção das pálpebras superior e inferior (telecante). Além disso, a condição pode estar associada a aberturas anormalmente baixas dos dutos lacrimais e uma maior suscetibilidade a infecções dos sacos lacrimais (dacriocistite). (Cada canto interno da fissura palpebral se abre em um pequeno espaço que contém uma abertura para o canal lacrimal.) os pacientes podem ter uma ponte nasal alta e anormalmente larga e "asas" nasais subdesenvolvidas (hipoplasia das asas nasais), o que leva ao estreitamento narinas. Além disso, em alguns casos, as sobrancelhas podem ser excepcionalmente espessas e / ou crescer juntas (sinofreeze). Em casos raros, as pessoas afetadas também apresentam olhos arregalados (hipertelorismo ocular). Como mencionado anteriormente, os pesquisadores descreveram diferentes formas da doença com base em certos sintomas e dados genéticos específicos. O CB do tipo II difere do CB do tipo I pela ausência de um canal de TV.
Algumas pessoas com VS podem ter anormalidades faciais adicionais. Eles podem incluir:
- uma ponta do nariz anormalmente arredondada que pode estar ligeiramente voltada para cima;
- "suavidade" anormal do sulco vertical do lábio superior (sulco);
- lábios carnudos;
- leve protrusão da mandíbula (prognatismo da mandíbula).
Também houve vários relatos em que a doença foi associada a uma fenda palatina (heiloschisis) e / ou um sulco anormal no lábio superior (lábio leporino).
A síndrome de Waardenburg está frequentemente associada a distúrbios da pigmentação devido a uma deficiência do pigmento melanina. Algumas pessoas com essa condição apresentam envelhecimento do couro cabeludo e dos cílios (poliose) ao nascer, o que tende a desaparecer com a idade. Sobrancelhas, cílios e cabelos do couro cabeludo podem ficar grisalhos ou brancos prematuramente (começando na infância, adolescência ou início da idade adulta). Além disso, alguns pacientes têm áreas de pele de formato irregular sem pigmentação (leucoderma ou vitiligo), especialmente no rosto e nas mãos. O distúrbio também pode estar associado ao subdesenvolvimento (hipoplasia) das fibras do tecido conjuntivo que constituem a maior parte da área manchada de ambos os olhos (íris). Como resultado, os pacientes podem ter olhos anormalmente azuis pálidos ou diferenças na pigmentação da íris ou em diferentes regiões da mesma íris (heterocromia da íris). Por exemplo, a íris de um olho pode ser azul e a da outra íris de uma cor diferente, ou uma ou ambas as íris podem parecer excepcionalmente "salpicadas". Alguns relatórios sugerem que a heterocromia da íris pode ser mais comum na síndrome de Waardenburg tipo 2, enquanto enquanto a presença de cabelos grisalhos no couro cabeludo e áreas despigmentadas da pele é mais comum em pessoas com síndrome de Waardenburg 1 modelo.
Leia também:Hipofosfatasia
Algumas pessoas com DF também sofrem de surdez congênita. Essa deficiência auditiva parece ser o resultado de uma anormalidade ou ausência do órgão de Corti, uma estrutura dentro da passagem em espiral oca do ouvido interno (cóclea). O órgão de Corti contém minúsculas células ciliadas que convertem as vibrações sonoras em impulsos nervosos, que são então transmitidos através do nervo auditivo (nervo coclear vestibular) para o cérebro. Anomalias no órgão de Corti podem interferir na transmissão desses impulsos nervosos, resultando em deficiência auditiva (chamada perda auditiva neurossensorial ou perda auditiva coclear). Na maioria dos pacientes com o transtorno, a surdez neurossensorial congênita afeta ambas as orelhas (bilateral). No entanto, em casos raros, apenas um lado (unilateral) pode ser afetado.
Em alguns casos, as características da face, olhos e audição podem ocorrer em conjunto com malformações bilaterais dos braços e mãos (membros superiores). Esta forma do distúrbio, que é descrita como uma manifestação grave de CB1, às vezes é chamada de síndrome Waardenburg tipo 3 (CB3), síndrome de Klein-Waardenburg ou síndrome de Waardenburg com anomalias superiores membros. Os defeitos bilaterais podem incluir:
- subdesenvolvimento (hipoplasia) e encurtamento anormal dos membros superiores;
- Flexão anormal de certas articulações dos dedos em posições fixas (contraturas de flexão)
- fusão dos ossos do pulso;
- e / ou tecido ou fusão (sindactilia) de certos dedos.
Em alguns casos, outras anormalidades esqueléticas podem estar presentes, como ombro alto congênito (deformidade de Sprengel).
Uma quarta forma da síndrome de Waardenburg também foi descrita, na qual as principais características da VS estão associadas à doença de Hirschsprung. Esta forma do distúrbio pode ser chamada de CB4, síndrome de Waardenburg-Schach ou síndrome de Waardenburg-Hirschsprung. Doença de Hirschsprung (também conhecido como megacólon aganglionar) é um distúrbio gastrointestinal caracterizado pela ausência de certos corpos de células nervosas (gânglios) na parede do músculo liso em área do cólon. Como resultado, há uma ausência ou violação das contrações rítmicas involuntárias que impulsionam os alimentos ao longo do trato gastrointestinal (peristalse). Os sintomas e sinais associados podem incluir:
- acúmulo anormal de fezes no cólon;
- expansão do cólon sobre o segmento afetado (megacólon);
- inchaço (flatulência);
- vômito;
- falta de apetite (anorexia);
- incapacidade de crescer e ganhar peso na taxa esperada;
- e / ou outros desvios.
Foram relatados casos raros de CB4 em que os pacientes também apresentaram sintomas neurológicos devido a anormalidades no cérebro e na medula espinhal (sistema nervoso central). Nesses casos, as indicações adicionais incluíram:
- restrição de crescimento;
- tônus muscular anormalmente diminuído (hipotensão);
- contraturas e deformidades dos membros (artrogripose);
- e / ou outros desvios.
Causas
Mutações genéticas EDN3, EDNRB, MITF, PAX3, SNAI2 e SOX10 pode causar a síndrome de Waardenburg. Esses genes estão envolvidos na formação e desenvolvimento de vários tipos de células, incluindo células produtoras de pigmentos chamadas melanócitos. Os melanócitos produzem um pigmento chamado melanina, que contribui para a cor da pele, do cabelo e dos olhos e desempenha um papel essencial no funcionamento normal do ouvido interno. Mutações em qualquer um desses genes interrompem o desenvolvimento normal dos melanócitos, resultando em pigmentação anormal da pele, cabelo, olhos e problemas auditivos.
Leia também:Uma dor de garganta
Os tipos I e III da síndrome de Waardenburg são causados por mutações no gene PAX3. Mutações genéticas MITF ou SNAI2 pode causar a síndrome de Waardenburg tipo II.
Mutações em genes SOX10, EDN3 ou EDNRB pode causar a síndrome de Waardenburg tipo IV. Além do desenvolvimento dos melanócitos, esses genes são importantes para o desenvolvimento das células nervosas do cólon. Mutações em um desses genes levam à perda de audição, alterações de pigmentação e problemas intestinais associados à doença de Hirschsprung.
Em alguns casos, a causa genética da síndrome de Waardenburg não foi estabelecida.
Tipo de herança
A síndrome de Waardenburg é geralmente herdada em uma forma autossômica dominante, o que significa que uma cópia do gene alterado em cada célula é suficiente para que o distúrbio ocorra. Na maioria dos casos, a pessoa afetada tem um dos pais com a doença. Uma pequena porcentagem dos casos é resultado de novas mutações em um gene; esses casos ocorrem em pessoas sem história familiar da doença.
Em alguns casos de síndrome de Waardenburg tipo II e IV, um padrão de herança autossômica recessiva é encontrado, o que significa que ambas as cópias do gene em cada célula têm mutações. Na maioria das vezes, os pais de um paciente com uma condição autossômica recessiva carregam uma cópia do gene mutado, mas não apresentam sinais e sintomas da doença.
Populações afetadas
A Síndrome de Waardenburg (SV) leva o nome do pesquisador (Petrus J. Waardenburg), que descreveu a doença pela primeira vez com precisão em 1951. Desde então, pelo menos 1.400 casos foram relatados na literatura médica. Os dados disponíveis indicam que a doença pode ter uma incidência de cerca de 1 em 40.000 nascimentos e é responsável por 2 a 5 por cento dos casos de surdez congênita. A doença afeta homens e mulheres de forma relativamente igual.
Distúrbios sintomáticos
Os sintomas dos distúrbios a seguir podem ser semelhantes aos da síndrome de Waardenburg. As comparações podem ser úteis para o diagnóstico diferencial.
Existem vários distúrbios que podem ter certas características semelhantes às observadas na VS. Por exemplo, de acordo com os pesquisadores, tais distúrbios podem incluir:
- casos familiares de parciais albinismo e surdez;
- casos familiares de vitiligo e perda auditiva neurossensorial congênita / neurossensorial;
- uma condição conhecida como síndrome de Vogt-Koyanagi-Harada.
Este último pode ser caracterizado por doenças inflamatórias dos olhos; vitiligo; envelhecimento das sobrancelhas, cílios e cabelos do couro cabeludo (poliose); perda de cabelo (alopecia); uma condição na qual certos sons podem causar desconforto (disacusão); e / ou outros sintomas e sinais.
Além disso, algumas anomalias congênitas também podem estar associadas ao deslocamento lateral do canto interno do olho (isto é, telescópico); olhos grandes (hipertelorismo ocular); narinas estreitas; sobrancelhas excepcionalmente grossas que podem crescer juntas (sinofreeze); deficiência auditiva; malformações dos membros superiores; desordens digestivas; e / ou outras funções potencialmente associadas à síndrome de Waardenburg. No entanto, esses distúrbios costumam ser caracterizados por sintomas adicionais e distintos, sinais físicos ou outras características que podem ajudar a distingui-los da VS.
Diagnóstico
A síndrome de Waardenburg (SV) pode ser diagnosticada no nascimento ou na primeira infância (ou, em alguns casos, mais tarde na vida) com base em uma avaliação clínica minuciosa, identificação de sinais físicos característicos, uma história completa do paciente e de sua família, bem como diversas especialidades pesquisar. Por exemplo, em pacientes com suspeita de CO, a avaliação diagnóstica pode incluir o uso de um paquímetro para medir a distância entre os cantos internos dos olhos, os cantos externos dos olhos e as pupilas (interpupilar distância). (Um paquímetro é um instrumento com dois braços articulados, móveis e curvos, usado para medir a espessura ou o diâmetro.) Os pesquisadores observam que a identificação e a análise a combinação dessas medidas às vezes pode ser útil para confirmar a presença ou ausência de deslocamento lateral do canto interno do olho, o que pode indicar a presença de VS 1 modelo.
Testes diagnósticos adicionais podem ser realizados para ajudar a detectar ou caracterizar certas anormalidades potencialmente associadas à doença. Esses exames podem incluir exame com um microscópio iluminado para visualizar as estruturas internas do olho (exame com uma lâmpada de fenda); pesquisa auditiva especializada; e / ou técnicas de imagem avançadas, como para avaliar anomalias do ouvido interno, defeitos esqueléticos (por exemplo, observada em SV tipo 3), doença de Hirschsprung (por exemplo, observada em distúrbio tipo 4), e etc.. Por exemplo, os pesquisadores apontam que a tomografia computadorizada (TC) pode ajudar a caracterizar defeitos no ouvido interno que são responsáveis pela surdez neurossensorial congênita. (A tomografia computadorizada usa um computador e raios-X para criar um filme que mostra imagens em corte transversal de estruturas internas.) Em alguns casos a avaliação diagnóstica também pode incluir a remoção (biópsia) e exame microscópico de certas amostras de tecido, como uma biópsia retal, para confirmar a doença Hirschsprung. Em alguns casos, testes diagnósticos adicionais também podem ser recomendados.
Leia também:Doença dela
Tratamentos padrão
O tratamento para a síndrome de Waardenburg visa eliminar sintomas específicos que aparecem em cada pessoa. Esse tratamento pode exigir o esforço coordenado de uma equipe de profissionais médicos, como médicos especializados em doenças da pele (dermatologistas); oftalmologistas (oftalmologistas); especialistas em audição; médicos envolvidos no diagnóstico e tratamento de doenças do esqueleto, articulações, músculos e tecidos relacionados (ortopedistas); médicos especializados em doenças do aparelho digestivo (gastroenterologistas); fonoaudiólogos; fisioterapeutas; e / ou outros profissionais de saúde.
O reconhecimento precoce da perda auditiva neurossensorial pode desempenhar um papel importante para garantir uma intervenção imediata e cuidados de suporte adequados. Em alguns casos, os médicos podem recomendar o tratamento com um implante coclear - um dispositivo que em que eletrodos implantados no ouvido interno estimulam o nervo auditivo para enviar impulsos para cérebro. Além disso, a educação especial precoce pode ser recomendada para auxiliar no desenvolvimento da fala e de técnicas específicas. (por exemplo, linguagem de sinais, leitura labial, uso de ferramentas de comunicação, etc.) que podem facilitar comunicação.
Como as pessoas com anomalias pigmentadas da pele podem ter predisposição a queimaduras solares e ao risco de desenvolver câncer de pele, os médicos podem recomendar evitar luz solar direta, usar protetor solar com alto fator de proteção solar (FPS), usar óculos escuros e revestimentos protetores contra sol. (como chapéus, mangas compridas, calças, etc.) e outras medidas apropriadas. Pacientes com pigmentação da íris reduzida, deslocamento lateral dos cantos internos dos olhos e / ou outras anormalidades oculares comórbidas, os oftalmologistas também podem recomendar certas medidas de suporte. Isso pode incluir o uso de óculos de cores especiais ou lentes de contato (por exemplo, para reduzir a possível sensibilidade à luz (fotofobia)), medidas para prevenir ou tratar infecções ou outras medidas preventivas ou terapêuticas.
Em pessoas com anormalidades nos membros superiores, o tratamento pode incluir fisioterapia e várias técnicas ortopédicas, possivelmente incluindo cirurgia. Além disso, às vezes a cirurgia pode ser recomendada para tratar outras anormalidades que podem estar associadas à doença. Os procedimentos cirúrgicos específicos realizados dependerão da gravidade e localização das anormalidades anatômicas, sintomas associados e outros fatores.
Por exemplo, para pacientes com doença de Hirschsprung, o tratamento pode exigir a remoção da área afetada do intestino e a “reunificação” cirúrgica das áreas saudáveis do intestino. Em alguns casos, antes da correção cirúrgica da condição, o tratamento pode exigir a criação de uma saída artificial para o cólon através de uma abertura na parede abdominal (por exemplo, uma válvula temporária colostomia).
Os serviços de apoio adicionais que algumas vítimas podem considerar úteis incluem educação especial e / ou outros serviços médicos, sociais ou profissionais. O aconselhamento genético também beneficiará as pessoas afetadas e suas famílias. Outro tratamento para a síndrome de Waardenburg é sintomático e de suporte.
Previsão
A síndrome de Waardenburg não deve afetar a expectativa de vida. Normalmente, a doença não é acompanhada por nenhuma outra complicação além de perda auditiva, distúrbios da pigmentação ou doença de Hirschsprung, que afeta o intestino grosso. As características físicas causadas por esta doença permanecerão com a pessoa por toda a vida.