Lipoidóza (lysozomálna choroba ukladania lipidov): čo to je, príklady
Pozor! Informácie slúžia len na informačné účely a nie sú určené na diagnostiku a predpisovanie liečby. Vždy sa poraďte so svojím odborným lekárom!
Obsah
- Čo sú lyzozomálne ochorenia?
- Čo sú to lipidy?
- Ako sa zdedí lipoidóza?
- Typy a symptómy lyzozomálnych chorôb
Čo sú lyzozomálne ochorenia?
Choroby lyzozomálneho ukladania (lipoidóza), sú skupinou dedičných metabolických ochorení, pri ktorých sa v rôznych bunkách a tkanivách tela hromadia škodlivé množstvá tukových látok (lipidov).
Ľudia s týmito poruchami buď nevytvárajú dostatok jedného z enzýmov potrebných na rozklad (metabolizáciu) lipidov, alebo produkujú enzýmy, ktoré nefungujú správne.
Toto nadmerné hromadenie tuku môže časom viesť k trvalému poškodeniu buniek a tkanív, najmä v mozgu, periférnom nervovom systéme (nervy od miechy po zvyšok telo), pečeň, slezina a kostnej drene.
Čo sú to lipidy?
Lipidy sú látky podobné tuku, ktoré sú dôležitou súčasťou membrán vo vnútri buniek a medzi nimi a v myelínovom obale, ktorý pokrýva a chráni nervy. Lipidy zahŕňajú oleje, mastné kyseliny, vosky, steroidy (ako je cholesterol a estrogén) a ďalšie príbuzné zlúčeniny.
Tieto tukové látky sa prirodzene ukladajú v bunkách, orgánoch a tkanivách tela. Drobné telá v bunkách, nazývané lyzozómy, pravidelne premieňajú alebo metabolizujú lipidy a proteíny na menšie zložky, ktoré telu dodávajú energiu. Poruchy, pri ktorých intracelulárny materiál, ktorý sa nemôže metabolizovať, zostáva v lyzozómoch, sa nazývajú lyzozomálne ochorenia.
Okrem chorôb spojených s akumuláciou lipidov zahŕňajú ďalšie lyzozomálne choroby spojené s akumuláciou mukolipidózy, pri ktorých sa akumulujú bunky a tkanivá. nadmerné množstvo lipidov s naviazanými molekulami cukru, ako aj mukopolysacharidózy, v ktorých sa hromadí nadmerné množstvo veľkého komplexného cukru molekuly.
Ako sa dedia lipoidóza?
Lysozomálne ochorenia sa dedia od jedného alebo oboch rodičov, ktorí sú nositeľmi defektného génu, ktorý reguluje špecifický enzým metabolizujúci lipidy v triede buniek v tele. Môžu byť zdedené dvoma spôsobmi:
- Autozomálne recesívna dedičnosť nastáva, keď obaja rodičia nesú a odovzdávajú kópiu defektného génu, ale ani jeden z rodičov nie je postihnutý poruchou. Každé dieťa narodené týmto rodičom má 25-percentnú šancu, že zdedí obe kópie defektného génu, 50-percentnú pravdepodobnosť, že bude nosičom podobným jeho rodičom, a 25-percentná šanca, že nezdedí žiadnu kópiu chybného gén. Deti oboch pohlaví môžu byť dedené autozomálne recesívnym spôsobom.
- X-viazaná (alebo pohlavná) recesívna dedičnosť nastáva, keď matka nesie postihnutý gén na X chromozóme. Chromozómy X a Y sa podieľajú na určovaní pohlavia. Ženy majú dva chromozómy X, zatiaľ čo muži majú jeden chromozóm X a jeden chromozóm Y. Synovia prenášačiek majú 50-percentnú šancu, že zdedia a ovplyvnia poruchu, keďže synovia dostávajú jeden chromozóm X od matky a jeden chromozóm Y od otca. Dcéry majú 50-percentnú šancu, že zdedia postihnutý chromozóm X od svojej matky a sú prenášačkami alebo mierne postihnuté. Postihnutí muži neprenášajú chorobu na svojich synov, no ich dcéry budú prenášačkami a prenášačkami choroby.
Typy a symptómy lyzozomálnych chorôb
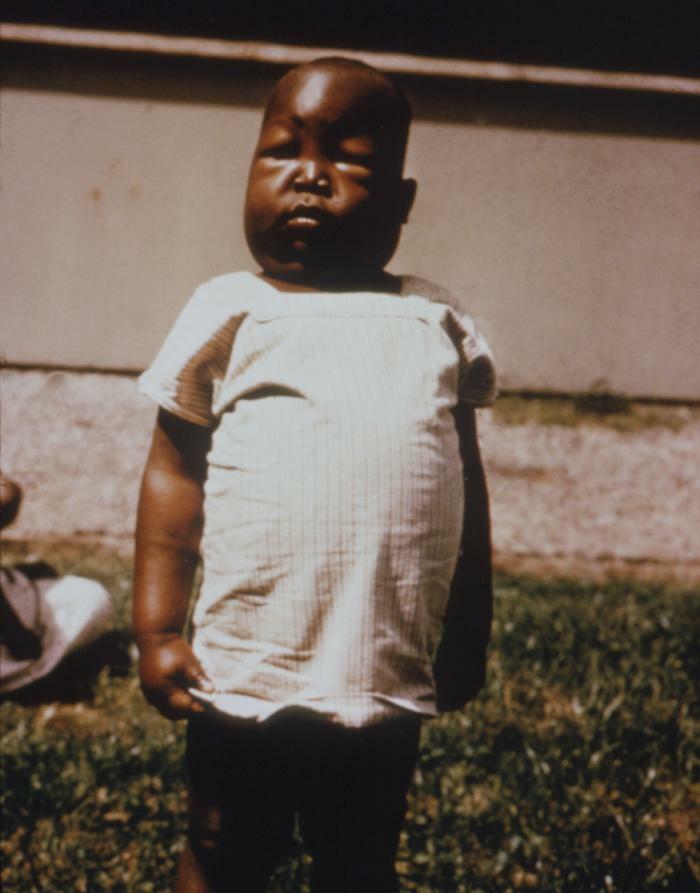
Gaucherova choroba - najčastejšie z lyzozomálnych ochorení. Ochorenie je spôsobené nedostatkom enzýmu glukocerebrosidázy, čo vedie k akumulácii glukocerebrozidu v mnohých tkanivách. Tukový materiál sa môže hromadiť v mozgu, slezine, pečeni, obličkách, pľúcach a kostnej dreni.
Symptómy môžu zahŕňať poškodenie mozgu, zväčšenie sleziny a pečene, zhoršená funkcia pečene, poruchy kostry a poškodenie kostí, ktoré môže spôsobiť bolesť a zlomeniny, opuch lymfatických uzlín a (niekedy) priľahlých kĺbov, nadúvanie, hnedastý tón pleti, anémia, nízky počet krvných doštičiek a žlté škvrny na očiach.
Najvážnejšie postihnutí jedinci môžu byť tiež náchylnejší na infekcie. Ochorenie postihuje rovnako mužov aj ženy.
Gaucherova choroba má tri všeobecné klinické podtypy:
- Typ 1 (Alebo nie neuronopatický typ) je najbežnejšou formou ochorenia v Spojených štátoch a Európe. Mozog nie je ovplyvnený, ale môže dôjsť k poškodeniu pľúc a v ojedinelých prípadoch aj obličiek. Symptómy sa môžu začať skoro v živote alebo v dospelosti a zahŕňajú zväčšenie pečene a závažné zväčšenie sleziny, čo môže viesť k prasknutiu a ďalším komplikáciám. Kostrová slabosť a ochorenie kostí môže byť rozsiahle. Ľuďom v tejto skupine sa zvyčajne ľahko tvoria modriny v dôsledku nízkeho počtu krvných doštičiek v krvi. Môžu tiež pociťovať únavu v dôsledku anémie. V závislosti od nástupu a závažnosti ochorenia môžu ľudia s typ 1 sa môže dožiť dospelosti. Mnohí pacienti majú mierne ochorenie alebo nemusia vykazovať žiadne príznaky. Predsa 1typu Gaucherova choroba je bežná medzi ľuďmi aškenázskeho židovského pôvodu a môže postihnúť ľudí akéhokoľvek etnického pôvodu.
- Typ 2 (alebo akútna detská neuropatická Gaucherova choroba) zvyčajne začína do 3 mesiacov po narodení. Symptómy zahŕňajú rozsiahle a progresívne poškodenie mozgu, spasticitu, kŕče, stuhnuté končatiny, zväčšenie pečene (hepatomegália pečene) a sleziny (splenomegália), abnormálny pohyb očí a slabá schopnosť sania a prehĺtania. Postihnuté deti zvyčajne zomierajú pred dosiahnutím veku 2 rokov.
- Typ 3 (chronický neuronopatický forma) môže začať kedykoľvek v detstve alebo aj v dospelosti. Je charakterizovaná pomaly progresívnymi, ale menej závažnými neurologickými symptómami v porovnaní s akútnou Gaucherovou chorobou 2 druhy. Medzi hlavné príznaky patria poruchy pohybu očí, kognitívne deficity, zlá koordinácia, záchvaty, zväčšenie sleziny a/alebo pečene, poruchy kostry, poruchy krvi vrátane anémie a problémy s dýchanie. Takmer každý s Gaucherovou chorobou 3 druhy, ktorý dostáva enzýmovú substitučnú liečbu, dosiahne dospelosť.
Pre pacientov 1. a 3. typ Enzýmová substitučná liečba, ktorá sa podáva intravenózne každé dva týždne, môže dramaticky znížiť veľkosť pečene a sleziny, znížiť abnormality kostry a zvrátiť ďalšie prejavy. Úspešná transplantácia kostnej drene lieči neurologické prejavy ochorenia. Tento postup však prináša značné riziká a u jedincov s Gaucherovou chorobou sa vykonáva len zriedka.
V zriedkavých prípadoch môže byť potrebný chirurgický zákrok na odstránenie celej sleziny alebo jej časti (ak má osoba veľmi nízky počet krvných doštičiek alebo ak zväčšený orgán výrazne ovplyvňuje pohodlie osoby). Krvná transfúzia môže byť prínosom pre niektorých ľudí s anémiou. Iní môžu potrebovať operáciu náhrady kĺbov na zlepšenie mobility a kvality života. V súčasnosti neexistuje účinná liečba poškodenia mozgu, ktoré sa môže vyskytnúť u ľudí s Gaucherovou chorobou typu 2 a 3.
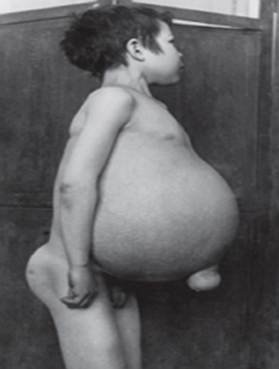
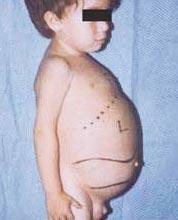
Niemann-Pickova choroba je skupina autozomálne recesívnych porúch spôsobených hromadením tuku a cholesterolu v bunkách pečene, sleziny, kostnej drene, pľúc a v niektorých prípadoch aj mozgu.
Neurologické komplikácie môžu zahŕňať ataxiu (nedostatok svalovej koordinácie, ktorá môže ovplyvniť chôdzu, písanie a jedenie, okrem iných funkcií), ochrnutie oka, degenerácia mozgu, problémy s učením, spasticita, ťažkosti s kŕmením a prehĺtaním, nezreteľná reč, strata svalového tonusu, precitlivenosť na dotyk a určitá zákal rohovky v dôsledku nadmerného hromadenia materiálov. U 50 % postihnutých sa okolo stredu sietnice vyvinie charakteristické čerešňovo-červené halo, ktoré môže vidieť lekár pomocou špeciálneho prístroja.
Niemann-Pickova choroba je rozdelená do troch všeobecných podtypov:
- Typ A, najťažšia forma, začína v ranom detstve. Bábätká po narodení vyzerajú normálne, ale do 6. mesiaca sa rozvinie hlboké poškodenie mozgu, zväčšená pečeň a slezina a opuchnuté lymfatické uzliny a uzliny pod kožou (xantómy). Slezina môže dosiahnuť 10-násobok svojej normálnej veľkosti a môže prasknúť, čo spôsobí krvácanie. Tieto deti postupne slabnú, strácajú motorické funkcie, môžu byť anemické a sú náchylné na opakované infekcie. Málokedy žijú dlhšie ako 18 mesiacov. Táto forma ochorenia sa najčastejšie vyskytuje v židovských rodinách.
- Typ B(alebo juvenilný nástup) zvyčajne neovplyvňuje mozog, ale väčšina detí sa vyvíja ataxiapoškodenie nervov opúšťajúcich miechu (periférna neuropatia) a pľúcne ťažkosti, ktoré progredujú s vekom. Pre dospievajúcich je typické zväčšenie pečene a sleziny. Ľudia s typ B môžu žiť relatívne dlho, ale mnohé vyžadujú celoživotnú oxygenoterapiu kvôli poškodeniu pľúc. Niemann-Pick typy A a B sú výsledkom akumulácie tukovej látky nazývanej sfingomyelín v dôsledku nedostatku enzýmu nazývaného sfingomyelináza.
- Typ C sa môže objaviť v ranom veku alebo sa môže vyvinúť počas dospievania alebo dokonca v dospelosti. Niemann-Pickova chorobatyp C nie je spôsobená nedostatkom sflingomyelinázy, ale nedostatkom proteínov NPC1 alebo NPC2. V dôsledku toho sa rôzne lipidy a najmä cholesterol hromadia v nervových bunkách a spôsobujú ich poruchu. Postihnutie mozgu môže byť rozsiahle, čo má za následok neschopnosť pozerať sa hore a dole, ťažkosti s chôdzou a prehĺtaním, progresívnu stratu sluchu a progresívnu demenciou. Ľudia s typ C majú len mierne zväčšenie sleziny a pečene. Stredná dĺžka života ľudí s typ C sa značne líši. Niektorí ľudia zomierajú v detstve, zatiaľ čo iní, ktorí sú menej postihnutí, môžu žiť aj v dospelosti.
V súčasnosti neexistuje žiadny liek na Niemann-Pickovu chorobu. Liečba je podporná. Deti zvyčajne zomierajú na infekciu alebo progresívnu neurologickú stratu. Vyskytli sa prípady transplantácie kostnej drene u niekoľkých pacientov s typ B so zmiešanými výsledkami.
Fabryho chorobataktiež známy ako nedostatok alfa-galaktozidázy-Aspôsobuje hromadenie tukových látok v autonómnom nervovom systéme (tej časti nervového systému, ktorá kontroluje mimovoľné funkcie, ako je dýchanie a srdcový tep), v očiach, obličkách a kardiovaskulárnom systéme systém.
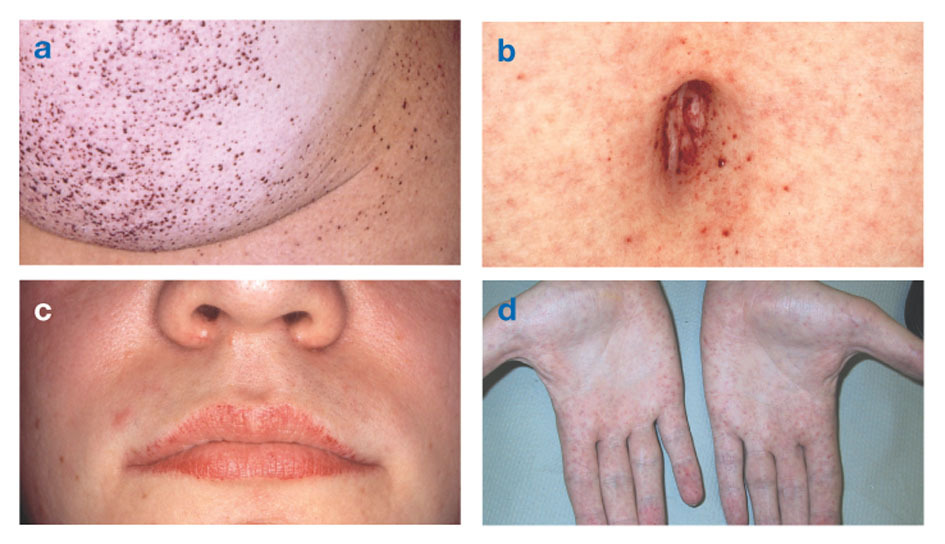
Fabryho choroba je jediné lipidové ochorenie viazané na X. Ochorenie postihuje najmä mužov, aj keď ľahšia a variabilnejšia forma sa vyskytuje u žien. Zriedkavo majú postihnuté ženy závažné symptómy podobné tým, ktoré sa vyskytujú u mužov s touto poruchou. Symptómy sa zvyčajne objavujú v detstve alebo dospievaní.
Neurologické príznaky zahŕňajú pálivú bolesť v rukách a nohách, ktorá sa zhoršuje v horúcom počasí alebo neskôr cvičenie, ako aj hromadenie prebytočného materiálu v priehľadných vrstvách rohovky (čo vedie k zakaleniu, ale bez zmeny videnia). Hromadenie tuku v stenách krvných ciev môže narušiť krvný obeh a vystaviť človeka riziku mŕtvica alebo infarkt. Medzi ďalšie príznaky patrí zväčšené srdce, progresívne zlyhanie obličiekčo vedie ku konečnému štádiu ochorenia obličiek, gastrointestinálnym problémom, zníženému poteniu a horúčke.
Angiokeratómy (malé, nerakovinové, červenofialové vyvýšené škvrny na koži) sa môžu vyvinúť v dolnej časti trupu a stávajú sa bežnejšími s vekom.
Ľudia s Fabryho chorobou často zomierajú predčasne na komplikácie zástava srdca, zlyhanie obličiek alebo mŕtvica. Lieky ako fenytoín a karbamazepín sa často predpisujú na liečbu bolesti, ktorá sprevádza Fabryho chorobu, ale nie na jej vyliečenie. Metoklopramid alebo Lipisorb (doplnok stravy) môžu zmierniť gastrointestinálne ťažkosti sa často vyskytuje u ľudí s Fabryho chorobou a niektorí ľudia môžu potrebovať transplantáciu obličky resp dialýza. Náhrada enzýmov môže u niektorých ľudí s Fabryho chorobou znížiť ukladanie, zmierniť bolesť a zachovať funkciu orgánov.
Farberova choroba taktiež známy ako Farberova lipogranulomatóza, opisuje skupinu zriedkavých autozomálne recesívnych porúch, ktoré spôsobujú hromadenie tukovej hmoty v kĺboch, tkanivách a centrálnom nervovom systéme. Postihuje mužov aj ženy. Ochorenie zvyčajne začína v ranom detstve, ale môže sa vyskytnúť aj neskôr v živote.
Rozvíjajú sa deti s klasickou Farberovou chorobou neurologické príznaky, ktoré môžu zahŕňať zvýšenú ospalosť a letargiu a problémy s prehĺtanie. Postihnutá môže byť aj pečeň, srdce a obličky. Ďalšie príznaky môžu zahŕňať kontraktúry kĺbov (chronické skrátenie svalov alebo šliach okolo kĺbov), vracanie, artritídu, zväčšenie lymfatické uzliny, zápal kĺbov, chrapot a hrčky pod kožou, ktoré sa pri postupe zhrubnú okolo kĺbov choroby.
Postihnutým ľuďom s dýchacími ťažkosťami môže byť potrebné zaviesť dýchaciu trubicu. Väčšina detí s týmto stavom zomiera do veku 2 rokov, zvyčajne od pľúcne ochorenia. Pri jednej z najťažších foriem ochorenia možno krátko po narodení diagnostikovať zväčšenie pečene a sleziny. Deti narodené s touto formou ochorenia zvyčajne zomierajú do 6 mesiacov.
Farberova choroba je spôsobená nedostatkom enzýmu nazývaného ceramidáza. V súčasnosti neexistuje žiadna špecifická liečba Farberovej choroby. Na zmiernenie bolesti môžu byť predpísané kortikosteroidy. Transplantácia kostnej drene môže znížiť výskyt granulómov (malé množstvo zapáleného tkaniva) u ľudí s miernym zápalom alebo u pacientov bez komplikácií pľúc alebo nervového systému. U starších ľudí možno granulómy zmenšiť alebo chirurgicky odstrániť.
Gangliosidóza pozostáva z dvoch rôznych skupín genetických chorôb. Obidve sú autozomálne recesívne a postihujú rovnako mužov aj ženy.
GM1 gangliozidóza.
Gangliozidóza GM1 je spôsobená nedostatkom enzýmu beta-galaktozidázy, čo vedie k abnormálnej akumulácii kyslé lipidové materiály, najmä v nervových bunkách centrálneho a periférneho nervového systému systémov. Gangliosidóza GM1 má tri klinické prejavy:
- GM1 (najzávažnejší podtyp, vyskytujúci sa krátko po narodení) môže zahŕňať neurodegeneráciu, záchvaty, zväčšenie pečene a sleziny, hrubé črty tváre, kostrové nepravidelnosti, stuhnutosť kĺbov, nadúvanie, svalová slabosť, prehnaná úľaková reakcia a problémy s chôdza. Asi u polovice postihnutých sa v oku objavia čerešňovočervené škvrny. Deti môžu byť hluché a slepé vo veku 1 a často zomierajú vo veku 3 rokov na srdcové komplikácie alebo zápal pľúc.
- Neskoré dieťa Gangliosidóza GM1 zvyčajne začína vo veku od 1 do 3 rokov. Neurologické symptómy zahŕňajú ataxiu, záchvaty, demenciu a ťažkosti s rozprávaním.
- GM1 gangliozidóza sa vyvíja vo veku od 3 do 30 rokov. Symptómy zahŕňajú zníženie svalovej hmoty (svalová atrofia), neurologické komplikácie, ktoré sú menej závažné a postupujú pomalšie ako iné formy poruchy, zákal rohovky u niektorých ľudí a trvalé svalové kontrakcie, ktoré spôsobujú krútenie a opakované pohyby alebo abnormálne držanie tela (dystónia). Angiokeratómy sa môžu vyvinúť v dolnej časti tela. Veľkosť pečene a sleziny je u väčšiny postihnutých ľudí normálna.
GM2 gangliozidóza.
GM2 gangliozidóza tiež spôsobuje, že telo hromadí prebytočné kyslé tukové látky v tkanivách a bunkách, predovšetkým nervových. Tieto poruchy sú výsledkom nedostatku enzýmu beta-hexosaminidázy. Poruchy GM2 zahŕňajú:
- Tay-Sachsova choroba (známa aj ako GM2 gangliozidóza variant B) a jej variantné formy sú spôsobené nedostatkom enzýmu hexosaminidázy A. Postihnuté deti sa počas prvých mesiacov života vyvíjajú normálne. Symptómy začínajú vo veku 6 mesiacov a zahŕňajú progresívnu stratu mentálnej kapacity, demenciu, znížený očný kontakt, zvýšený šoková reakcia na hluk, progresívna strata sluchu vedúca k hluchote, ťažkostiam s prehĺtaním, slepote, čerešňovým červeným škvrnám na sietnici a paralýza. Záchvaty môžu začať v druhom roku života dieťaťa. Bábätká sa môžu časom stať závislými na kŕmnej trubici a často zomierajú vo veku 4 rokov na opakujúce sa infekcie. Žiaľ, neexistuje žiadna špeciálna liečba. Antikonvulzíva môžu spočiatku kontrolovať záchvaty. Medzi ďalšie podporné liečby patrí správna výživa a hydratácia a metódy na udržanie otvorených dýchacích ciest. Zriedkavá forma poruchy tzv neskorým nástupom Tay-Sachsovej chorobysa vyskytuje u ľudí vo veku 20 až 30 rokov a vyznačuje sa nestabilnou chôdzou a progresívnym neurologickým zhoršovaním.
- Sandhoffova choroba (Možnosť AB) je ťažká forma Tay-Sachsovej choroby. Nástup zvyčajne nastáva vo veku 6 mesiacov a nie je obmedzený na žiadnu etnickú skupinu. Neurologické príznaky môžu zahŕňať progresívne zhoršovanie centrálneho nervového systému, motora slabosť, skorá slepota, ťažká vystrašená reakcia na zvuk, spasticita, šok alebo zášklby svaly (myoklonus), epilepsiaabnormálne zväčšená hlava (makrocefália) a červené škvrny v oku. Ďalšie príznaky môžu zahŕňať časté infekcie dýchacích ciest, srdcový šelest, bábikovité črty tváre a zväčšenie pečene a sleziny. Neexistuje žiadna špecifická liečba Sandhoffovej choroby. Rovnako ako pri Tay-Sachsovej chorobe, podporná starostlivosť zahŕňa neustále udržiavanie dýchacích ciest otvorených, ako aj správnu výživu a hydratáciu. Antikonvulzíva môžu spočiatku kontrolovať epilepsiu (záchvaty).
Krabbeho choroba(taktiež známy ako globulárna bunková leukodystrofia a galaktozylceramidová lipidóza) je autozomálne recesívne ochorenie spôsobené nedostatkom enzýmu galaktocerebrozidázy. Ochorenie najčastejšie postihuje dojčatá s nástupom pred dosiahnutím veku 6 mesiacov, ale môže sa vyskytnúť počas dospievania alebo dospelosti.
Hromadenie nestrávených tukov ovplyvňuje rast ochranného obalu nervu (myelínového obalu) a spôsobuje vážne poškodenie mentálnych a motorických schopností. Medzi ďalšie príznaky patrí svalová slabosť, znížená schopnosť svalov natiahnuť sa (hypotónia), svalové napätie (spasticita), náhly šok alebo zášklby končatín (myoklonické záchvaty), podráždenosť, nevysvetliteľná horúčka, hluchota, slepota, paralýza a ťažkosti s prehĺtaním. Môže dôjsť aj k dlhodobému úbytku hmotnosti.
Ochorenie možno diagnostikovať testovaním enzýmov a identifikáciou jeho charakteristického bunkového zoskupenia do globoidných teliesok v bielej hmote mozgu, demyelinizácia nervov a degenerácia a deštrukcia mozgových buniek. U dojčiat je ochorenie zvyčajne smrteľné pred dosiahnutím veku 2 rokov. Ľudia s pokročilejšou formou ochorenia majú miernejší priebeh ochorenia a žijú podstatne dlhšie.
Pre Krabbeho chorobu nebola vyvinutá žiadna špecifická liečba, aj keď včasná transplantácia kostnej drene môže niektorým ľuďom pomôcť. Ľudia s pokročilejšou formou ochorenia majú miernejší priebeh ochorenia a žijú podstatne dlhšie.
Metachromatická leukodystrofiaalebo MLD, je skupina porúch charakterizovaných hromadením tukových látok v bielej hmote centrálneho nervového systému a v periférnych nervoch a do určitej miery v obličkách. Podobne ako Krabbeho choroba, MLD ovplyvňuje myelín, ktorý pokrýva a chráni nervy. Toto autozomálne recesívne ochorenie je spôsobené nedostatkom enzýmu arylsulfatázy A. Táto porucha postihuje mužov aj ženy.
Metachromatická leukodystrofia má tri charakteristické formy: neskorú dojčenskú, juvenilnú a dospelú.
- Neskorá detská MLD zvyčajne začína medzi 12 a 20 mesiacmi po narodení. Deti sa môžu na prvý pohľad zdať normálne, ale majú ťažkosti s chôdzou a tendenciou padať, sprevádzané prerušovanou bolesťou rúk a nôh. progresívna strata zraku vedúca k slepote, oneskoreniu vo vývoji a strate predtým získaných míľnikov, zhoršenému prehĺtaniu, záchvatom a demencii do 2 rokov. U detí sa tiež rozvíja progresívne ochabovanie svalov a slabosť a nakoniec strácajú schopnosť chodiť. Väčšina detí s touto formou poruchy zomiera do veku 5 rokov.
- Juvenilná MLD zvyčajne začína vo veku od 3 do 10 rokov. Symptómy zahŕňajú zhoršený školský výkon, mentálne postihnutie, ataxiu, záchvaty a demenciu. Symptómy progredujú, pričom smrť nastáva 10–20 rokov po nástupe ochorenia.
- Forma pre dospelých. Symptómy dospelých nástup po 16. roku života a môže zahŕňať ataxiu, záchvaty, abnormálne chvenie končatín (tras), poruchy koncentrácie, depresie, duševné poruchy a demencia. Smrť zvyčajne nastáva 6-14 rokov po nástupe symptómov.
MLD sa nedá vyliečiť. Liečba je symptomatická a podporná. Transplantácia kostnej drene môže v niektorých prípadoch oddialiť progresiu ochorenia. Významný pokrok sa dosiahol s génovou terapiou na zvieracích modeloch s MLD a v klinických štúdiách.
Wolmanova chorobataktiež známy ako nedostatok lyzozomálnej kyslej lipázyje závažná porucha ukladania lipidov, ktorá je zvyčajne smrteľná vo veku 1 roka. Toto autozomálne recesívne ochorenie je charakterizované akumuláciou esterov cholesterolu (zvyčajne transportná forma cholesterolu) a triglyceridy (chemická forma, v ktorej existujú tuky v tele), ktoré sa môžu výrazne hromadiť a spôsobiť poškodenie buniek a tkaniny.
Touto poruchou trpia muži aj ženy. Dojčatá sú normálne a aktívne pri narodení, ale rýchlo sa u nich rozvinie progresívna mentálna porucha, zväčšená pečeň a výrazne zväčšená slezina, nadúvanie, gastrointestinálne problémy. žltačka, anémia, vracanie a usadzovanie vápnika v nadobličkyspôsobí ich stvrdnutie.
Iný typ nedostatok lyzozomálnej kyslej lipázy je choroba z ukladania esterov cholesterolu. Toto extrémne zriedkavé ochorenie vzniká v dôsledku ukladania esterov cholesterolu a triglyceridov v krvinkáchlymfatické a lymfoidné tkanivo. U detí v dospelosti sa pečeň zväčšuje, čo vedie k cirhóze a chronický zlyhanie pečene. Deti môžu mať aj usadeniny vápnika v nadobličkách a v neskorších štádiách ochorenia sa môže vyvinúť žltačka.
V súčasnosti sa problematika náhrady enzýmov aktívne študuje tak pri Wolmanovej chorobe, ako aj pri akumulácii esteru cholesterolu.
Pozor! Informácie slúžia len na informačné účely a nie sú určené na diagnostiku a predpisovanie liečby. Vždy sa poraďte so svojím odborným lekárom!