Waardenburgov sindrom: kaj je to, simptomi, zdravljenje, prognoza
Vsebina
- Kaj je Waardenburgov sindrom?
- Znaki in simptomi
- Vzroki
- Vrsta dedovanja
- Prizadete populacije
- Simptomatske motnje
- Diagnostika
- Standardno zdravljenje
- Napoved
Kaj je Waardenburgov sindrom?
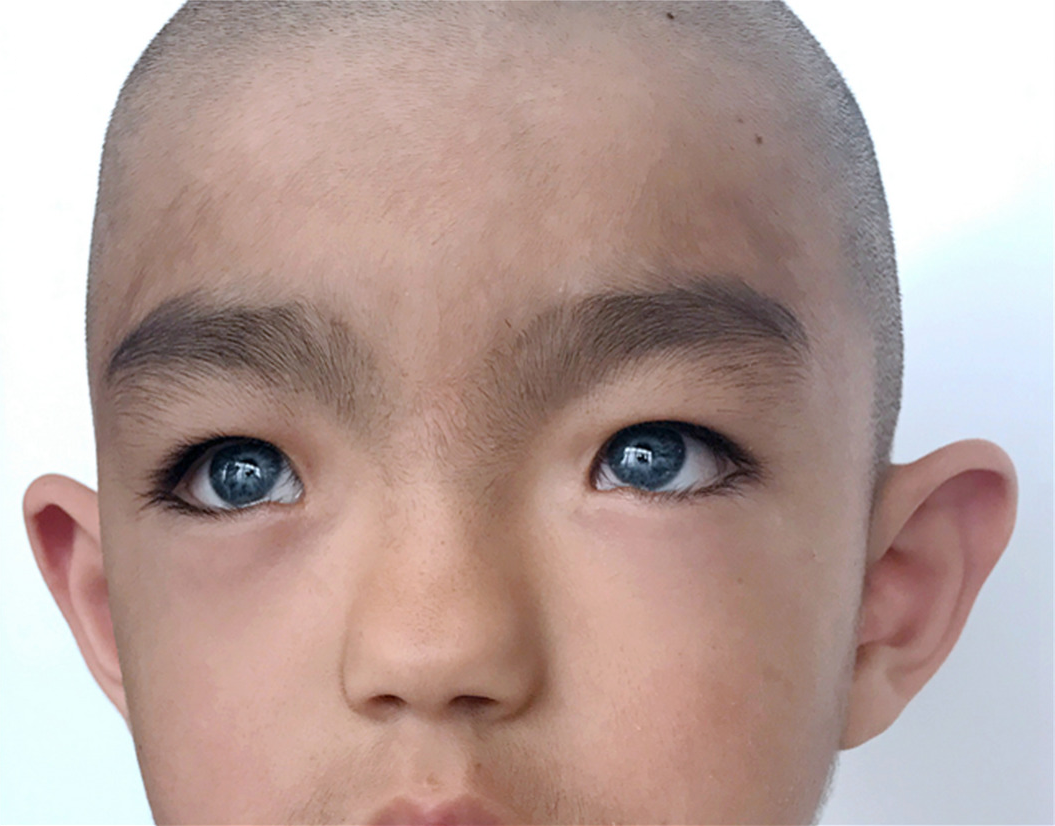
Waardenburgov sindrom Je genetska motnja, ki se lahko pojavi ob rojstvu (prirojena). Obseg in resnost pridruženih simptomov in znakov se lahko od primera do primera zelo razlikujeta. Vendar pa primarni znaki pogosto vključujejo značilne nepravilnosti obraza; nenavadno zmanjšano obarvanje (pigmentacija) las, kože in / ali šarenice obeh očes; in / ali prirojena gluhost. Natančneje, nekateri bolniki imajo lahko zaradi stranskega (stranskega) premika notranjih kotičkov oči (telekant) nenavadno širok nos. Poleg tega lahko nepravilnosti pri pigmentaciji vključujejo bel pramen las, ki raste nad čelom; prezgodnje sivenje ali beljenje las; razlike v barvi dveh šarenic ali v različnih delih iste šarenice (heterokromija šarenice); in / ali neenakomerno svetla (depigmentirana) področja kože (levkoderma). Nekateri ljudje imajo lahko tudi okvaro sluha zaradi nenormalnosti v notranjem ušesu (senzorinevralna izguba sluha).
Raziskovalci so opisali različne vrste Waardenburgovega sindroma (v nadaljevanju skr. SV) na podlagi povezanih simptomov in specifičnih genetskih podatkov. Na primer, Waardenburgov sindrom tipa I (CB1) je običajno povezan s stranskim premikom notranjega očesnega kota (t.j. teleskopski), vendar tip II (CB2) ni povezan s to značilnostjo. Poleg tega je znano, da sta CB1 in CB2 posledica sprememb (mutacij) v različnih genih. Druga oblika, znana kot tip III (CB3), pri kateri so značilne lastnosti obraza, oči (okular) in motnje sluha lahko povezane z značilnimi malformacijami rok in rok (zgornjih okončin). Četrta oblika, znana kot CB4 ali Waardenburg-Hirschsprungova bolezen, ima lahko primarne znake SV v kombinaciji z Hirschsprungova bolezen. Slednji je prebavna (gastrointestinalna) motnja, pri kateri ni skupine specializiranih teles živčnih celic v gladki (neprostovoljni) mišični steni debeline črevesja.
V večini primerov se Waardenburgov sindrom prenaša avtosomno dominantno. Ugotovljeno je bilo več različnih genov bolezni, ki lahko pri nekaterih posameznikih ali družinah (sorodnikih) povzročijo Waardenburgov sindrom.
Znaki in simptomi
Primarni znaki Waardenburgovega sindroma so lahko:
- značilne obrazne anomalije (glej. Fotografija);
- motnje pigmentacije (hipopigmentacija) las, kože in / ali šarenice ali šarenice obeh oči (delno) albinizem);
- prirojena izguba sluha.
Kot smo že omenili, so lahko povezani simptomi in znaki zelo različni, tudi med prizadetimi družinskimi člani. Na primer, medtem ko imajo nekateri bolniki lahko samo eno značilnost, imajo drugi lahko več nenormalnosti, povezanih z boleznijo.
Pri nekaterih bolnikih z Waardenburgovim sindromom (SV) pride do nenormalnega premika stranskih (stranskih) notranjih kotičkov oči, ki jih tvori stik zgornje in spodnje veke (telekant). Poleg tega je lahko stanje povezano z nenavadno nizkimi odprtinami solznih kanalov in povečano dovzetnostjo za okužbe solznih vrečk (dakriocistitis). (Vsak notranji kot palpebralne razpoke se odpre v majhen prostor, ki vsebuje odprtino za solzni kanal.) bolniki imajo lahko nenormalno širok, visok nosni most in nerazvita nosna "krila" (hipoplazija nosnih kril), kar vodi v ozko nosnice. Poleg tega so lahko v nekaterih primerih obrvi nenavadno debele in / ali zrastejo skupaj (sinofriz). V redkih primerih imajo prizadeti ljudje tudi široko razmaknjene oči (očesni hipertelorizem). Kot smo že omenili, so raziskovalci na podlagi določenih simptomov in specifičnih genetskih podatkov opisali različne oblike bolezni. CB tipa II se od CB tipa I razlikuje po odsotnosti televizijskega kanala.
Nekateri ljudje s SV imajo lahko dodatne nepravilnosti na obrazu. Lahko vključujejo:
- nenavadno zaobljen vrh nosu, ki je lahko rahlo obrnjen navzgor;
- nenormalna "gladkost" navpičnega utora zgornje ustnice (utor);
- polne ustnice;
- rahla izboklina spodnje čeljusti (prognatizem spodnje čeljusti).
Bilo je tudi več poročil, v katerih je bila bolezen povezana z razcepom neba (heiloschisis) in / ali nenormalen utor na zgornji ustnici (razcepljena ustnica).
Waardenburgov sindrom je pogosto povezan s pigmentacijskimi motnjami zaradi pomanjkanja pigmenta melanina. Nekateri s tem stanjem imajo ob rojstvu sivenje lasišča in trepalnic (polioza), ki s starostjo ponavadi izgine. Obrvi, trepalnice in lasje lasišča lahko predčasno postanejo sivi ali beli (od zgodnjega otroštva, mladosti ali zgodnje odraslosti). Poleg tega imajo nekateri bolniki nepravilne oblike kože brez pigmentacije (levkoderma ali vitiligo), zlasti na obrazu in rokah. Motnja je lahko povezana tudi s nerazvitostjo (hipoplazijo) vezivnega tkiva, ki sestavljajo večino obarvanega območja obeh očes (šarenice). Posledično imajo lahko bolniki nenavadno bledo modre oči ali razlike v pigmentaciji šarenice ali v različnih regijah iste šarenice (šarenica heterokromija). Na primer, šarenica enega očesa je lahko modra, druge šarenice pa drugačne barve, ali pa se lahko ena ali obe šarenici zdijo nenavadno "pegaste". Nekatera poročila kažejo, da je heterokromija šarenice pogostejša pri Waardenburgovem sindromu tipa 2, medtem ko medtem ko je prisotnost sivih las na lasišču in depigmentiranih predelih kože pogostejša pri ljudeh z Waardenburgovim sindromom 1 tip.
Preberite tudi:Hipofosfatazija
Nekateri ljudje s FD trpijo tudi zaradi prirojene gluhost. Zdi se, da je ta okvara sluha posledica nenormalnosti ali odsotnosti Cortijevega organa, strukture znotraj votlega spiralnega prehoda notranjega ušesa (polža). Cortijev organ vsebuje drobne lasne celice, ki pretvarjajo zvočne vibracije v živčne impulze, ki se nato po slušnem živcu (vestibularni kohlearni živec) prenašajo v možgane. Anomalije v Cortijevem organu lahko motijo prenos takšnih živčnih impulzov, kar povzroči okvaro sluha (imenovano senzorinevralna izguba sluha ali kohlearna izguba sluha). Pri večini bolnikov z motnjo prirojena senzorinevralna gluhost prizadene obe ušesi (obojestransko). V redkih primerih pa je lahko prizadeta le ena stran (enostranska).
V nekaterih primerih se lahko pojavijo značilne lastnosti obraza, oči in sluha v povezavi z dvostranskimi malformacijami rok in rok (zgornjih okončin). Ta oblika motnje, ki je opisana kot huda manifestacija CB1, se včasih imenuje sindrom Waardenburgov tip 3 (CB3), Klein-Waardenburgov sindrom ali Waardenburgov sindrom z zgornjim okončine. Dvostranske napake lahko vključujejo:
- nerazvitost (hipoplazija) in nenormalno skrajšanje zgornjih okončin;
- Nenormalno upogibanje nekaterih sklepov prstov v fiksnih položajih (upogibne kontrakture)
- zlitje kosti zapestja;
- in / ali trakovi ali zlitje (sindaktilija) določenih prstov.
V nekaterih primerih so lahko prisotne tudi druge skeletne nepravilnosti, na primer prirojeno visoko ramensko stojanje (Sprengelova deformacija).
Opisana je bila tudi četrta oblika Waardenburgovega sindroma, pri kateri so glavne značilnosti SV povezane s Hirschsprungovo boleznijo. Ta oblika motnje se lahko imenuje CB4, Waardenburg-Schachov sindrom ali Waardenburg-Hirschsprungov sindrom. Hirschsprungova bolezen (znan tudi kot aganglionski megakolon) je prebavna motnja označena z odsotnostjo določenih teles živčnih celic (ganglijev) v gladki mišični steni v območje debelega črevesa. Posledica tega je odsotnost ali kršitev neprostovoljnih ritmičnih krčev, ki hrano poganjajo vzdolž prebavil (peristaltika). Povezani simptomi in znaki lahko vključujejo:
- nenormalno kopičenje blata v debelem črevesu;
- razširitev debelega črevesa na prizadeti segment (megakolon);
- napihnjenost (napenjanje);
- bruhanje;
- pomanjkanje apetita (anoreksija);
- nezmožnost rasti in pridobivanja teže po pričakovani stopnji;
- in / ali druga odstopanja.
Poročali so o redkih primerih CB4, pri katerih so imeli bolniki tudi nevrološke simptome zaradi nenormalnosti v možganih in hrbtenjači (centralni živčni sistem). V takih primerih so dodatne indikacije vključevale:
- omejitev rasti;
- nenormalno zmanjšan mišični tonus (hipotenzija);
- kontrakture in deformacije okončin (artrogripoza);
- in / ali druga odstopanja.
Vzroki
Genske mutacije EDN3, EDNRB, MITF, PAX3, SNAI2 in SOX10 lahko povzroči Waardenburgov sindrom. Ti geni so vključeni v nastanek in razvoj več vrst celic, vključno s celicami, ki proizvajajo pigmente, imenovane melanociti. Melanociti proizvajajo pigment, imenovan melanin, ki prispeva k barvi kože, las in oči ter ima bistveno vlogo pri normalnem delovanju notranjega ušesa. Mutacije katerega koli od teh genov motijo normalen razvoj melanocitov, kar povzroči nenormalno pigmentacijo kože, las, oči in težave s sluhom.
Preberite tudi:Vneto grlo
Tipa I in III Waardenburgovega sindroma povzročajo mutacije v genu PAX3. Genske mutacije MITF ali SNAI2 lahko povzroči Waardenburgov sindrom tipa II.
Mutacije v geni SOX10, EDN3 ali EDNRB lahko povzroči Waardenburgov sindrom tipa IV. Poleg razvoja melanocitov so ti geni pomembni za razvoj živčnih celic v debelem črevesu. Mutacije v enem od teh genov povzročajo izgubo sluha, spremembe pigmentacije in črevesne težave, povezane s Hirschsprungovo boleznijo.
V nekaterih primerih genetski vzrok Waardenburgovega sindroma ni bil ugotovljen.
Vrsta dedovanja
Waardenburgov sindrom se običajno podeduje v avtosomno dominantni obliki, kar pomeni, da je za pojav motnje dovolj ena kopija spremenjenega gena v vsaki celici. V večini primerov ima prizadeta oseba enega od staršev s tem stanjem. Majhen odstotek primerov je posledica novih mutacij v genu; takšni primeri se pojavijo pri ljudeh, ki nimajo družinske anamneze bolezni.
V nekaterih primerih Waardenburgovega sindroma tipa II in IV najdemo avtosomno recesivni dedni vzorec, kar pomeni, da imata obe kopiji gena v vsaki celici mutacije. Najpogosteje starši bolnika z avtosomno recesivnim stanjem nosijo eno kopijo mutiranega gena, vendar ne kažejo znakov in simptomov bolezni.
Prizadete populacije
Waardenburgov sindrom (SV) je dobil ime po raziskovalcu (Petrus J. Waardenburg), ki je prvič natančno opisal motnjo leta 1951. Od takrat je bilo v medicinski literaturi zabeleženih najmanj 1400 primerov. Razpoložljivi podatki kažejo, da je lahko pojavnost bolezni približno 1 na 40.000 rojstev in je odgovorna za 2 do 5 odstotkov primerov prirojene gluhost. Bolezen prizadene moške in ženske relativno enako.
Simptomatske motnje
Simptomi naslednjih motenj so lahko podobni simptomom Waardenburgovega sindroma. Primerjave so lahko koristne za diferencialno diagnozo.
Obstajajo številne motnje, ki imajo lahko določene lastnosti, podobne tistim pri SV. Na primer, po mnenju raziskovalcev lahko takšne motnje vključujejo:
- družinski primeri delnih albinizem in gluhost;
- družinski primeri vitiliga in prirojene senzorinevralne / senzorinevralne izgube sluha;
- stanje znano kot sindrom Vogt-Koyanagi-Harada.
Za slednje so lahko značilne vnetne očesne bolezni; vitiligo; sivenje obrvi, trepalnic in lasišča (polioza); izguba las (alopecija); stanje, v katerem lahko določeni zvoki povzročijo nelagodje (diskusija); in / ali drugih simptomov in znakov.
Poleg tega so lahko nekatere prirojene nepravilnosti povezane tudi s stranskim premikom notranjega očesnega kota (t.j. teleskopskim); široko postavljene oči (očesni hipertelorizem); ozke nosnice; nenavadno debele obrvi, ki lahko rastejo skupaj (sinofriz); okvara sluha; malformacije zgornjih okončin; prebavne motnje; in / ali druge funkcije, ki so potencialno povezane z Waardenburgovim sindromom. Vendar pa so za take motnje pogosto značilni dodatni, značilni simptomi, fizični znaki ali druge značilnosti, ki jih lahko razlikujejo od SV.
Diagnostika
Waardenburgov sindrom (SV) je mogoče diagnosticirati ob rojstvu ali zgodnjem otroštvu (ali v nekaterih primerih kasneje v življenju) na podlagi temeljito klinično oceno, identifikacijo značilnih telesnih znakov, popolno anamnezo bolnika in njegove družine ter različne specializirane raziskave. Na primer, pri bolnikih s sumom na CO lahko diagnostična ocena vključuje uporabo čeljusti za merjenje razdalje med notranjimi kotički oči, zunanjimi kotički oči in zenicami (interpupilarna razdalja). (Čeljust je instrument z dvema zgibnima, premičnima, ukrivljenima rokama, ki se uporablja za merjenje debeline ali premera.) Raziskovalci ugotavljajo, da identifikacija in analiza kombinacija teh meritev je včasih lahko koristna za potrditev prisotnosti ali odsotnosti stranskega premika notranjega očesnega kota, kar lahko kaže na prisotnost SV 1 tip.
Za odkrivanje ali opredelitev določenih nepravilnosti, ki so potencialno povezane z boleznijo, se lahko izvedejo dodatni diagnostični testi. Takšni pregledi lahko vključujejo pregled z osvetljenim mikroskopom za vizualizacijo notranjih struktur očesa (pregled z razrezano svetilko); specializirane slušne raziskave; in / ali napredne tehnike slikanja, na primer za ocenjevanje anomalij notranjega ušesa, okvar okostja (na primer pri SV tipa 3), Hirschsprungovi bolezni (npr. pri motnji tipa 4) in itd.. Raziskovalci na primer opozarjajo, da lahko računalniška tomografija (CT) pomaga pri opredelitvi napak v notranjem ušesu, ki so odgovorne za prirojeno senzorinevralno gluhost. (Računalniška tomografija uporablja računalnik in rentgenske žarke za ustvarjanje filma, ki prikazuje slike prečnih prerezov notranjih struktur.) V nekaterih primerih diagnostična ocena lahko vključuje tudi odstranitev (biopsijo) in mikroskopski pregled nekaterih vzorcev tkiva, kot je rektalna biopsija, za potrditev bolezni Hirschsprung. V nekaterih primerih se lahko priporočijo tudi dodatni diagnostični testi.
Preberite tudi:Njena bolezen
Standardno zdravljenje
Zdravljenje Waardenburgovega sindroma je namenjeno odpravljanju posebnih simptomov, ki se pojavijo pri vsaki osebi. Takšno zdravljenje lahko zahteva usklajena prizadevanja ekipe zdravstvenih delavcev, na primer zdravnikov, specializiranih za kožna stanja (dermatologi); oftalmologi (oftalmologi); strokovnjaki za sluh; zdravniki, ki sodelujejo pri diagnosticiranju in zdravljenju bolezni okostja, sklepov, mišic in sorodnih tkiv (ortopedi); zdravniki, specializirani za bolezni prebavnega trakta (gastroenterologi); logopedi; fizioterapevti; in / ali drugih zdravstvenih delavcev.
Zgodnje prepoznavanje senzorinevralne izgube sluha ima lahko pomembno vlogo pri zagotavljanju takojšnje intervencije in ustrezne podporne oskrbe. V nekaterih primerih lahko zdravniki priporočijo zdravljenje s kohlearnim vsadkom - napravo, ki pri katerem elektrode, implantirane v notranje uho, stimulirajo slušni živec, da pošilja impulze možgane. Poleg tega se lahko priporoči zgodnje posebno izobraževanje za pomoč pri razvoju govora in posebnih tehnik. (na primer znakovni jezik, branje ustnic, uporaba komunikacijskih orodij itd.), ki lahko olajšajo komunikacijo.
Ker so ljudje s pigmentiranimi kožnimi nepravilnostmi lahko nagnjeni k sončnim opeklinam in tveganju za razvoj kožnega raka, lahko zdravniki priporočijo izogibajte se neposredni sončni svetlobi, uporabljajte kremo za sončenje z visokim faktorjem zaščite pred soncem (SPF), nosite sončna očala in zaščitne premaze proti sonce. (kot so klobuki, dolgi rokavi, hlače itd.) in drugi ustrezni ukrepi. Bolniki z zmanjšano pigmentacijo šarenice, bočnim premikom notranjih kotičkov oči in / ali druge komorbidne očesne nepravilnosti, lahko tudi oftalmologi priporočijo nekatere podporne ukrepe. To lahko vključuje uporabo posebej zatemnjenih očal ali kontaktnih leč (na primer za zmanjšanje možne občutljivosti na svetlobo (fotofobija)), ukrepe za preprečevanje ali zdravljenje okužbe ali druge preventivne ali terapevtske ukrepe.
Pri ljudeh z nepravilnostmi zgornjih okončin lahko zdravljenje vključuje fizikalno terapijo in različne ortopedske tehnike, po možnosti tudi operacijo. Poleg tega se včasih lahko priporoči operacija za zdravljenje drugih nepravilnosti, ki so lahko povezane z boleznijo. Posebni kirurški posegi bodo odvisni od resnosti in lokacije anatomskih nepravilnosti, povezanih simptomov in drugih dejavnikov.
Na primer, pri bolnikih s Hirschsprungovo boleznijo bo zdravljenje morda zahtevalo odstranitev prizadetega dela črevesja in kirurško "ponovno združitev" zdravih črevesnih območij. V nekaterih primerih lahko pred kirurško korekcijo stanja zdravljenje zahteva oblikovanje umetno odprtino za debelo črevo skozi odprtino v trebušni steni (na primer začasno) kolostomija).
Dodatne podporne storitve, ki so nekaterim žrtvam v pomoč, vključujejo posebno izobraževanje in / ali druge zdravstvene, socialne ali strokovne storitve. Genetsko svetovanje bo koristilo tudi prizadetim in njihovim družinam. Drugo zdravljenje Waardenburgovega sindroma je simptomatsko in podporno.
Napoved
Waardenburgov sindrom ne bi smel vplivati na pričakovano življenjsko dobo. Običajno bolezni ne spremljajo drugi zapleti, razen izgube sluha, motenj pigmentacije ali Hirschsprungove bolezni, ki prizadene debelo črevo. Fizične lastnosti, ki jih povzroča ta bolezen, bodo pri človeku ostale vse življenje.