
Lipoidoos (lüsosomaalne lipiidide ladestumise haigus): mis see on, näited
Tähelepanu! Teave on ainult informatiivsel eesmärgil ega ole mõeldud diagnoosimiseks ega ravi määramiseks. Konsulteerige alati oma eriarstiga!
Sisu
- Mis on lüsosomaalsed ladustamishaigused?
- Mis on lipiidid?
- Kuidas lipoidoosi päritakse?
- Lüsosomaalsete ladestushaiguste tüübid ja sümptomid
Mis on lüsosomaalsed ladustamishaigused?
Lüsosomaalsed ladestushaigused (lipoidoos), on rühm pärilikke ainevahetushaigusi, mille puhul keha erinevatesse rakkudesse ja kudedesse koguneb kahjulikes kogustes rasvaineid (lipiide).
Nende häiretega inimesed kas ei tooda piisavalt üht lipiidide lagundamiseks (metaboliseerimiseks) vajalikku ensüümi või toodavad ensüüme, mis ei tööta korralikult.
Aja jooksul võib see liigne rasva kogunemine põhjustada rakkude ja kudede püsivaid kahjustusi, eriti ajus, perifeerses närvisüsteemis (närvid seljaajust kuni ülejäänud keha), maks, põrn ja luuüdi.
Mis on lipiidid?
Lipiidid on rasvataolised ained, mis on membraanide olulised osad rakkude sees ja vahel ning müeliinkestas, mis katab ja kaitseb närve. Lipiidide hulka kuuluvad õlid, rasvhapped, vahad, steroidid (nagu kolesterool ja östrogeen) ja muud sarnased ühendid.
Neid rasvaineid säilitatakse loomulikult keha rakkudes, elundites ja kudedes. Rakkude väikesed kehad, mida nimetatakse lüsosoomideks, muudavad või metaboliseerivad regulaarselt lipiide ja valke väiksemateks komponentideks, et anda kehale energiat. Häireid, mille korral rakusisene materjal, mida ei saa metaboliseerida, jääb lüsosoomidesse, nimetatakse lüsosomaalseteks ladestushaigusteks.
Lisaks lipiidide kogunemisega seotud haigustele hõlmavad muud akumulatsiooniga seotud lüsosomaalsed haigused mukolipidoosid, mille käigus rakud ja koed akumuleeruvad. liigne kogus lipiide, millele on kinnitatud suhkrumolekulid, samuti mukopolüsahharidoosid, millesse koguneb liigne kogus suurt keerulist suhkrut molekulid.
Kuidas päritakse lipoidoos?
Lüsosomaalsed säilitushaigused on päritud ühelt või mõlemalt vanemalt, kellel on defektne geen, mis reguleerib teatud lipiide metaboliseerivat ensüümi teatud keharakkude klassis. Neid saab pärida kahel viisil:
- Autosoomne retsessiivne pärand tekib siis, kui mõlemad vanemad kannavad ja annavad edasi defektse geeni koopiat, kuid häire ei mõjuta kumbagi vanemat. Igal nendele vanematele sündinud lapsel on 25-protsendiline võimalus pärida mõlemad defektse geeni koopiad, 50 protsenti. tõenäosus, et ta on oma vanematega sarnane kandja, ja 25-protsendiline tõenäosus, et ta ei päri ühtegi defektse koopiat geen. Mõlemast soost lapsed võivad pärida autosoomselt retsessiivselt.
- X-seotud (või sooga seotud) retsessiivne pärand tekib siis, kui ema kannab mõjutatud geeni X-kromosoomis. X- ja Y-kromosoomid osalevad soo määramisel. Naistel on kaks X-kromosoomi, meestel aga üks X-kromosoom ja üks Y-kromosoom. Naissoost kandjate poegadel on 50-protsendiline tõenäosus haigust pärida ja mõjutada, kuna pojad saavad ühe X-kromosoomi emalt ja ühe Y-kromosoomi isalt. Tütardel on 50-protsendiline tõenäosus, et nad pärivad kahjustatud X-kromosoomi oma emalt ja nad on kandjad või kergelt haiged. Haigestunud mehed ei edasta haigust oma poegadele, kuid nende tütred on selle haiguse kandjad ja edasikandjad.
Lüsosomaalsete ladestushaiguste tüübid ja sümptomid
Gaucher' tõbi - lüsosomaalsetest haigustest kõige levinum. Haiguse põhjuseks on ensüümi glükotserebrosidaasi puudulikkus, mis põhjustab glükotserebrosiidi akumuleerumist paljudes kudedes. Rasvmaterjal võib koguneda ajju, põrna, maksa, neerudesse, kopsudesse ja luuüdi.
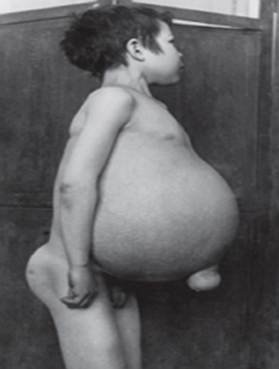
Sümptomiteks võivad olla ajukahjustus, põrna suurenemine ja maks, maksafunktsiooni kahjustus, luustiku häired ja luukahjustused, mis võivad põhjustada valu ja luumurde, lümfisõlmede ja (mõnikord) külgnevate liigeste turset, puhitus, pruunikas nahatoon, aneemia, madal trombotsüütide arv ja kollased laigud silmadel.
Kõige tõsisemalt mõjutatud isikud võivad olla ka nakkustele vastuvõtlikumad. Haigus mõjutab võrdselt mehi ja naisi.
Gaucher' tõvel on kolm üldist kliinilist alatüüpi:
- Tüüp 1 (Või ei neuronopaatiline tüüp) on selle haiguse kõige levinum vorm Ameerika Ühendriikides ja Euroopas. Aju ei mõjuta, kuid võib esineda kopsude ja harvadel juhtudel ka neerude kahjustusi. Sümptomid võivad alata varases eas või täiskasvanueas ning hõlmata maksa suurenemist ja põrna tõsist suurenemist, mis võib põhjustada rebenemist ja täiendavaid tüsistusi. Skeleti nõrkus ja luuhaigused võivad olla ulatuslikud. Sellesse rühma kuuluvatel inimestel tekivad tavaliselt kergesti verevalumid, kuna nende veres on madal trombotsüütide arv. Samuti võivad nad kogeda aneemia tõttu väsimust. Olenevalt haiguse algusest ja raskusastmest võivad inimesed, kellel tüüp 1 võib elada täiskasvanuks. Paljudel haigetel on kerge haigus või neil ei pruugi olla mingeid sümptomeid. Kuigi 1tüüp Gaucheri tõbi on levinud Ashkenazi juudi päritolu inimeste seas ja võib mõjutada igasuguse etnilise taustaga inimesi.
- Tüüp 2 (või äge lapsepõlve neuropaatiline Gaucher' tõbi) algab tavaliselt 3 kuu jooksul pärast sündi. Sümptomiteks on ulatuslik ja progresseeruv ajukahjustus, spastilisus, krambid, jäigad jäsemed, maksa suurenemine (maksa hepatomegaalia) ja põrn (splenomegaalia), silmade ebanormaalne liikumine ning halb imemis- ja neelamisvõime. Mõjutatud lapsed surevad tavaliselt enne 2-aastaseks saamist.
- Tüüp 3 (krooniline neuronopaatiline vorm) võib alata igal ajal lapsepõlves või isegi täiskasvanueas. Seda iseloomustavad aeglaselt progresseeruvad, kuid vähem rasked neuroloogilised sümptomid võrreldes ägeda Gaucher' tõvega 2 tüüpi. Peamised sümptomid on silmade liikumise häired, kognitiivsed häired, halb koordinatsioon, krambid, põrna ja/või maksa suurenemine, luustiku häired, verehaigused, sealhulgas aneemia, ja probleemid hingamine. Peaaegu kõik, kellel on Gaucher' tõbi 3 tüüpi, kes saab ensüümasendusravi, jõuab täiskasvanuikka.
Patsientide jaoks 1. ja 3. tüüp Ensüüm-asendusravi, mida manustatakse intravenoosselt iga kahe nädala tagant, võib järsult vähendada maksa ja põrna suurust, vähendada luustiku kõrvalekaldeid ja muuta teisi ilminguid. Eduka luuüdi siirdamisega ravitakse haiguse neuroloogilisi ilminguid. Selle protseduuriga kaasnevad aga märkimisväärsed riskid ja seda tehakse Gaucher' tõbe põdevatel inimestel harva.
Harvadel juhtudel võib kogu põrna või selle osa eemaldamiseks olla vajalik operatsioon (kui inimesel on väga madal trombotsüütide arv või kui suurenenud elund mõjutab oluliselt inimese mugavust). Vereülekanne võib olla kasulik mõnele aneemiaga inimesele. Teised võivad vajada liigeseasendusoperatsiooni, et parandada liikuvust ja elukvaliteeti. Praegu puudub tõhus ravi 2. ja 3. tüüpi Gaucher' tõvega inimestel tekkida võiva ajukahjustuse jaoks.
Niemann-Picki tõbi on autosoomsete retsessiivsete häirete rühm, mis on põhjustatud rasva ja kolesterooli kogunemisest maksa, põrna, luuüdi, kopsude ja mõnel juhul ka aju rakkudesse.
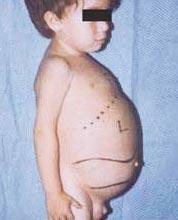
Neuroloogiliste tüsistuste hulka võivad kuuluda ataksia (lihaste koordinatsiooni puudumine, mis võib mõjutada kõndimist, kirjutamist ja söömist, muude funktsioonide hulgas), silmade halvatus, aju degeneratsioon, õpiprobleemid, spastilisus, toitumis- ja neelamisraskused, hägune kõne, lihastoonuse kaotus, ülitundlikkus puudutuse suhtes ja sarvkesta mõningane hägusus liigsest kogunemisest materjalid. 50% haigestunud inimestest tekib võrkkesta keskkoha ümber iseloomulik kirsipunane halo, mida arst saab näha spetsiaalse instrumendiga.
Niemann-Picki tõbi jaguneb kolmeks üldiseks alatüübiks:
- Tüüp A, kõige raskem vorm, algab varases lapsepõlves. Imikud tunduvad sündides normaalsed, kuid 6 kuu pärast tekib sügav ajukahjustus, maks ja põrn ning lümfisõlmede ja nahaaluste sõlmede turse (ksantoomid). Põrn võib muutuda 10 korda tavapärasest suuremaks ja rebeneda, põhjustades verejooksu. Need lapsed muutuvad järk-järgult nõrgemaks, kaotavad motoorsed funktsioonid, võivad muutuda aneemiliseks ja neil on korduvad infektsioonid. Nad elavad harva üle 18 kuu. Seda haigusvormi esineb kõige sagedamini juudi perekondades.
- Tüüp B(või alaealine algus) ei mõjuta tavaliselt aju, kuid enamik lapsi areneb ataksia, seljaajust lahkuvate närvide kahjustus (perifeerne neuropaatia) ja kopsuprobleemid, mis edenevad koos vanusega. Maksa ja põrna suurenemine on tüüpiline noorukitele. Inimesed, kellel on tüüp B võib elada suhteliselt kaua, kuid paljud vajavad kopsukahjustuse tõttu elukestvat hapnikravi. Niemann-Picki tüübid A ja B on sfingomüelinaasi nimelise ensüümi puudulikkuse tõttu akumuleeruvad rasvained, mida nimetatakse sfingomüeliiniks.
- Tüüp C võib ilmneda varases eas või areneda noorukieas või isegi täiskasvanueas. Niemann-Picki tõbitüüp C ei ole põhjustatud sflingomüelinaasi puudulikkusest, vaid NPC1 või NPC2 valkude puudumisest. Selle tulemusena kogunevad närvirakkudesse erinevad lipiidid ja eriti kolesterool, mis põhjustab nende talitlushäireid. Aju haaratus võib olla ulatuslik, mille tulemuseks on võimetus üles-alla vaadata, kõndimis- ja neelamisraskused, progresseeruv kuulmislangus ja progresseeruv. dementsus. Inimesed, kellel on tüüp C põrna ja maksa suurenemine on kerge. Inimeste eeldatav eluiga tüüp C varieerub tunduvalt. Mõned inimesed surevad lapsepõlves, samas kui teised, keda see mõjutab vähem, võivad elada ka täiskasvanueas.
Niemann-Picki tõbe ei ole praegu ravitud. Ravi on toetav. Lapsed surevad tavaliselt infektsiooni või progresseeruva neuroloogilise kaotuse tõttu. Mitmetel patsientidel on esinenud luuüdi siirdamist tüüp B segaste tulemustega.
Fabry haigustuntud ka kui alfa-galaktosidaas-A puudulikkus, põhjustab rasvainete kogunemist autonoomses närvisüsteemis (närvisüsteemi selles osas, mis kontrollib tahtmatuid funktsioone, nagu hingamine ja südamelöök), silmades, neerudes ja südame-veresoonkonnas süsteem.
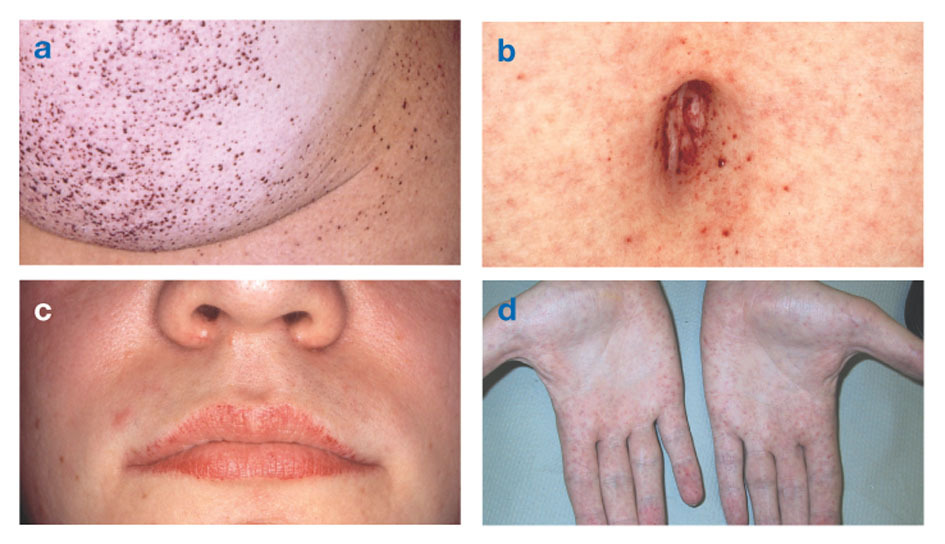
Fabry tõbi on ainus X-seotud lipiidide haigus. See haigus mõjutab peamiselt mehi, kuigi naistel esineb kergem ja varieeruvam vorm. Harva esinevad haigetel naistel tõsised sümptomid, mis on sarnased selle häirega meeste omadega. Sümptomid ilmnevad tavaliselt lapsepõlves või noorukieas.
Neuroloogiliste nähtude hulka kuuluvad põletav valu kätes ja jalgades, mis süveneb kuuma ilmaga või pärast seda treeningut, samuti liigse materjali kogunemist sarvkesta läbipaistvatesse kihtidesse (mis põhjustab hägusust, kuid ilma muutused nägemises). Rasva kogunemine veresoonte seintesse võib häirida vereringet, seades inimese ohtu insult või südameatakk. Teised sümptomid hõlmavad südame suurenemist, progresseeruvat neerupuudulikkusmis põhjustab lõppstaadiumis neeruhaigust, seedetrakti probleeme, higistamise vähenemist ja palavikku.
Angiokeratoomid (väikesed, mittevähilised, punakaslillad kõrgenenud laigud nahal) võivad areneda torso alaosas ja sageneda vanusega.
Fabry tõbe põdevad inimesed surevad sageli tüsistuste tõttu enneaegselt südamepuudulikkus, neerupuudulikkus või insult. Selliseid ravimeid nagu fenütoiin ja karbamasepiin kirjutatakse sageli välja Fabry tõvega kaasneva valu leevendamiseks, kuid mitte selle raviks. Metoklopramiid või Lipisorb (toidulisand) võivad leevendada seedetrakti häireid esineb sageli Fabry tõbe põdevatel inimestel ja mõned inimesed võivad vajada neerusiirdamist või dialüüs. Ensüümide asendamine võib mõnel Fabry tõvega inimesel vähendada ladustamist, leevendada valu ja säilitada elundite funktsiooni.
Farberi haigus tuntud ka kui Farberi lipogranulomatoos, kirjeldab haruldaste autosoomsete retsessiivsete häirete rühma, mis põhjustavad rasvainete kogunemist liigestesse, kudedesse ja kesknärvisüsteemi. See mõjutab nii mehi kui naisi. Haigus algab tavaliselt varases lapsepõlves, kuid võib tekkida ka hilisemas elus.
Klassikalise Farberi tõvega lapsed arenevad neuroloogilised sümptomid, mille hulka võivad kuuluda suurenenud unisus ja letargia ning probleemid neelamine. Samuti võivad olla kahjustatud maks, süda ja neerud. Teised sümptomid võivad hõlmata liigeste kontraktuure (liigeseid ümbritsevate lihaste või kõõluste krooniline lühenemine), oksendamist, artriiti, laienemist. lümfisõlmed, liigesepõletik, häälekähedus ja nahaalused tükid, mis edenedes liigeste ümber paksenevad haigused.
Hingamisraskustega haigetel võib olla vaja sisestada hingamistoru. Enamik selle haigusseisundiga lapsi sureb 2-aastaselt, tavaliselt alates kopsuhaigused. Haiguse ühe raskeima vormi korral võib kohe pärast sündi diagnoosida suurenenud maks ja põrn. Selle haigusvormiga sündinud lapsed surevad tavaliselt 6 kuu jooksul.
Farberi tõbi on põhjustatud ensüümi keramidaasi puudulikkusest. Praegu puudub Farberi tõve spetsiifiline ravi. Valu leevendamiseks võib välja kirjutada kortikosteroide. Luuüdi siirdamine võib vähendada granuloomide (põletikuliste kudede väikeste masside) arvu kerge põletikuga inimestel või patsientidel, kellel ei ole kopsu- või närvisüsteemi tüsistusi. Vanematel inimestel saab granuloome kahandada või kirurgiliselt eemaldada.
Gangliosidoos koosneb kahest erinevast geneetiliste haiguste rühmast. Mõlemad on autosoomselt retsessiivsed ja mõjutavad võrdselt nii mehi kui ka naisi.
GM1 gangliosidoos.
GM1 gangliosidoos on põhjustatud ensüümi beeta-galaktosidaasi puudulikkusest, mille tulemuseks on ebanormaalne kuhjumine happelised lipiidmaterjalid, eriti kesk- ja perifeerse närvisüsteemi närvirakkudes süsteemid. GM1 gangliosidoosil on kolm kliinilist ilmingut:
- GM1 (kõige raskem alatüüp, mis esineb varsti pärast sündi) võib hõlmata neurodegeneratsiooni, krampe, maksa ja põrna suurenemist, näojoonte jämedus, luustiku ebakorrapärasused, liigeste jäikus, puhitus, lihasnõrkus, liialdatud ehmatusreaktsioon ja probleemid kõnnak. Umbes pooltel haigetest tekivad silma kirsipunased laigud. Lapsed võivad 1-aastaselt olla kurdid ja pimedad ning sageli surra 3-aastaselt südametüsistustesse või kopsupõletik.
- Hiline laps GM1 gangliosidoos algab tavaliselt vanuses 1 kuni 3 aastat. Neuroloogiliste sümptomite hulka kuuluvad ataksia, krambid, dementsus ja kõneraskused.
- GM1 gangliosidoos areneb vanuses 3 kuni 30 aastat. Sümptomiteks on lihasmassi vähenemine (lihaste atroofia), neuroloogilised tüsistused, mis on vähem rasked ja arenevad aeglasemalt kui muud vormid häired, mõnel inimesel sarvkesta hägusus ja püsivad lihaskontraktsioonid, mis põhjustavad keerdumist ja korduvaid liigutusi või ebanormaalseid kehaasendeid (düstoonia). Angiokeratoomid võivad tekkida kehatüve alumises osas. Maksa ja põrna suurus on enamikul haigetel inimestel normaalne.
GM2 gangliosidoos.
GM2 gangliosidoos põhjustab ka liigsete happeliste rasvainete kogunemist kudedesse ja rakkudesse, eelkõige närvirakkudesse. Need häired tulenevad ensüümi beeta-heksosaminidaasi puudulikkusest. GM2 häired hõlmavad:
- Tay-Sachsi haigus (tuntud ka kui GM2 gangliosidoosi variant B) ja selle teisendvormid on põhjustatud ensüümi heksosaminidaas A puudulikkusest. Mõjutatud lapsed arenevad normaalselt esimestel elukuudel. Sümptomid algavad 6 kuu vanuselt ja nende hulka kuuluvad progresseeruv vaimse võimekuse kaotus, dementsus, silmside vähenemine, suurenenud ehmatav reaktsioon mürale, progresseeruv kuulmislangus, mis põhjustab kurtust, neelamisraskused, pimedus, kirsipunased laigud võrkkestal ja mõned halvatus. Krambid võivad alata lapse teisel eluaastal. Imikud võivad lõpuks muutuda toitmissondist sõltuvaks ja surevad sageli korduvate infektsioonide tõttu 4-aastaselt. Erilist ravi kahjuks ei ole. Antikonvulsandid võivad esialgu krampe kontrollida. Teised toetavad ravimeetodid hõlmavad õiget toitumist ja hüdratatsiooni ning hingamisteede avatuna hoidmise meetodeid. Häire haruldane vorm, mida nimetatakse hilise algusega Tay-Sachsi tõbi, esineb 20–30-aastastel inimestel ja seda iseloomustab ebastabiilne kõnnak ja progresseeruv neuroloogiline halvenemine.
- Sandhoffi tõbi (valik AB) on Tay-Sachsi tõve raske vorm. Algab tavaliselt 6 kuu vanuselt ja see ei piirdu ühegi etnilise rühmaga. Neuroloogiliste tunnuste hulka võivad kuuluda kesknärvisüsteemi progresseeruv halvenemine, motoorne nõrkus, varajane pimedus, tugev hirmunud reaktsioon helile, spastilisus, šokk või tõmblused lihased (müokloonus), epilepsia, ebanormaalselt suurenenud pea (makrotsefaalia) ja punased laigud silmas. Teised sümptomid võivad hõlmata sagedasi hingamisteede infektsioone, südame mühin, nukulaadsed näojooned ning maksa ja põrna suurenemine. Sandhoffi tõve jaoks puudub spetsiifiline ravi. Nagu Tay-Sachsi tõve puhul, hõlmab toetav ravi hingamisteede kogu aeg lahtihoidmist, samuti õiget toitumist ja hüdratatsiooni. Antikonvulsandid suudavad esialgu kontrollida epilepsiat (krampe).
Krabbe haigus(tuntud ka kui globulaarsete rakkude leukodüstroofia ja galaktosüültseramiidi lipidoos) on autosoomne retsessiivne haigus, mis on põhjustatud ensüümi galaktotserebrosidaasi puudulikkusest. See haigus mõjutab kõige sagedamini imikuid, kes algavad enne 6 kuu vanust, kuid võib esineda ka noorukieas või täiskasvanueas.
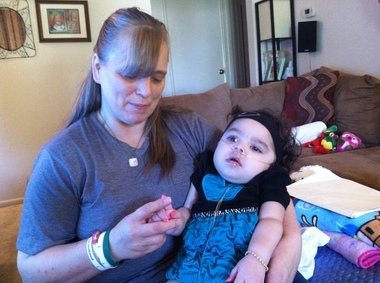
Seedimata rasvade kogunemine mõjutab närvide kaitsekesta (müeliinkesta) kasvu ning põhjustab vaimsete ja motoorsete oskuste tõsist kahjustust. Teised sümptomid on lihasnõrkus, lihaste venitusvõime vähenemine (hüpotoonia), lihaspinge (spastilisus), äkiline šokk. või jäsemete tõmblused (müokloonilised krambid), ärrituvus, seletamatu palavik, kurtus, pimedus, halvatus ja neelamisraskused. Võib tekkida ka pikaajaline kaalulangus.
Haigust saab diagnoosida ensüümide testimise ja sellele iseloomuliku rakurühma tuvastamise teel globoidkehadeks aju valgeaines, närvide demüeliniseerumiseks ja degeneratsiooniks ning ajurakkude hävimiseks. Imikutel lõppeb haigus tavaliselt surmaga enne 2. eluaastat. Kaugelearenenud haigusvormiga inimestel on haiguse kulg kergem ja nad elavad oluliselt kauem.
Krabbe tõve spetsiifilist ravi ei ole välja töötatud, kuigi varajane luuüdi siirdamine võib mõnda inimest aidata. Kaugelearenenud haigusvormiga inimestel on haiguse kulg kergem ja nad elavad oluliselt kauem.
Metakromaatiline leukodüstroofiaMLD ehk MLD on rühm häireid, mida iseloomustab rasvainete kuhjumine kesknärvisüsteemi valgeaines ja perifeersetes närvides ning teatud määral ka neerudes. Sarnaselt Krabbe tõvega mõjutab MLD müeliini, mis katab ja kaitseb närve. See autosoomne retsessiivne häire on põhjustatud ensüümi arüülsulfataasi A puudulikkusest. See häire mõjutab nii mehi kui naisi.
Metakromaatilisel leukodüstroofial on kolm iseloomulikku vormi: hiline imik, alaealine ja täiskasvanu.
- Hiline imikute MLD algab tavaliselt 12–20 kuud pärast sündi. Lapsed võivad alguses tunduda normaalsed, kuid neil on raskusi kõndimisel ja kalduvus kukkuda, millega kaasneb vahelduv valu kätes ja jalgades. progresseeruv nägemise kaotus, mis põhjustab pimedust, arengupeetust ja varem omandatud verstapostide kaotust, neelamishäireid, krampe ja dementsust kuni 2 aastat. Lastel areneb ka progresseeruv lihaste kurnatus ja nõrkus ning lõpuks kaotavad nad kõndimisvõime. Enamik selle häire vormiga lapsi sureb 5-aastaselt.
- Alaealiste MLD algab tavaliselt 3–10 aasta vanuselt. Sümptomiteks on halvenenud koolisooritus, vaimne kahjustus, ataksia, krambid ja dementsus. Sümptomid progresseeruvad, surm saabub 10–20 aastat pärast haiguse algust.
- Täiskasvanu vorm. Sümptomid täiskasvanud algab pärast 16. eluaastat ja võib hõlmata ataksiat, krampe, jäsemete ebanormaalset värinat (treemor), keskendumishäireid, depressioon, psüühikahäired ja dementsus. Surm saabub tavaliselt 6–14 aastat pärast sümptomite tekkimist.
MLD-d ei saa ravida. Ravi on sümptomaatiline ja toetav. Luuüdi siirdamine võib mõnel juhul haiguse progresseerumist edasi lükata. MLD-ga loommudelites ja kliinilistes uuringutes on geeniteraapiaga tehtud olulisi edusamme.
Wolmani haigustuntud ka kui lüsosomaalse happe lipaasi puudulikkuson tõsine lipiidide ladestumise häire, mis tavaliselt lõppeb surmaga 1-aastaselt. Seda autosoomset retsessiivset häiret iseloomustab kolesterooli estrite (tavaliselt kolesterooli transpordivorm) kuhjumine ja triglütseriidid (keemiline vorm, milles rasvad kehas eksisteerivad), mis võivad märkimisväärselt akumuleeruda ja põhjustada rakukahjustusi ja kangad.
Selle haiguse all kannatavad nii mehed kui naised. Imikud on sündides normaalsed ja aktiivsed, kuid neil areneb kiiresti progresseeruv vaimne kahjustus, suurenenud maks ja oluliselt suurenenud põrn, kõhupuhitus, seedetrakti probleemid. kollatõbi, aneemia, oksendamine ja kaltsiumi ladestumine neerupealisedpõhjustades nende kõvenemist.
Teine tüüp lüsosomaalse happe lipaasi puudulikkus on kolesterooli estri ladustamise haigus. See üliharuldane haigus tekib kolesterooli estrite ja triglütseriidid vererakkudes, lümfi- ja lümfoidkoest. Täiskasvanueas lastel maks suureneb, mis põhjustab tsirroosi ja krooniline maksapuudulikkus. Lastel võib neerupealistes olla ka kaltsiumi ladestumist, haiguse hilisemates staadiumides võib tekkida kollatõbi.
Praegu uuritakse aktiivselt ensüümide asendamise küsimust nii Wolmani tõve kui ka kolesterooli estri akumulatsiooni puhul.
Tähelepanu! Teave on ainult informatiivsel eesmärgil ega ole mõeldud diagnoosimiseks ega ravi määramiseks. Konsulteerige alati oma eriarstiga!


