
Lipoidoza (lizosomalna bolest skladištenja lipida): što je to, primjeri
Pažnja! Informacije su samo u informativne svrhe i nisu namijenjene postavljanju dijagnoze i propisivanju liječenja. Uvijek se posavjetujte sa svojim liječnikom specijalistom!
Sadržaj
- Što su lizosomske bolesti skladištenja?
- Što su lipidi?
- Kako se lipoidoza nasljeđuje?
- Vrste i simptomi lizosomskih bolesti skladištenja
Što su lizosomske bolesti skladištenja?
Lizosomalne bolesti skladištenja (lipoidoza), skupina su nasljednih metaboličkih bolesti kod kojih se u različitim stanicama i tkivima tijela nakupljaju štetne količine masnih tvari (lipida).
Osobe s ovim poremećajima ili ne proizvode dovoljno jednog od enzima potrebnih za razgradnju (metabolizaciju) lipida ili proizvode enzime koji ne rade ispravno.
S vremenom, ovo prekomjerno nakupljanje masti može dovesti do trajnog oštećenja stanica i tkiva, osobito u mozgu, perifernom živčanom sustavu (živci od leđne moždine do ostatka tijelo), jetra, slezena i koštane srži.
Što su lipidi?
Lipidi su tvari slične mastima koje su važni dijelovi membrana unutar i između stanica te u mijelinskoj ovojnici, koja prekriva i štiti živce. Lipidi uključuju ulja, masne kiseline, voskove, steroide (kao što su kolesterol i estrogen) i druge srodne spojeve.
Ove masne tvari prirodno su pohranjene u stanicama, organima i tkivima tijela. Sitna tijela unutar stanica, nazvana lizosomi, redovito pretvaraju ili metaboliziraju lipide i proteine u manje komponente kako bi tijelu dali energiju. Poremećaji u kojima unutarstanični materijal, koji se ne može metabolizirati, ostaje u lizosomima, nazivaju se lizosomske bolesti skladištenja.
Osim bolesti povezanih s nakupljanjem lipida, druge lizosomske bolesti povezane s nakupljanjem uključuju mukolipidoze, u kojima se nakupljaju stanice i tkiva prekomjerna količina lipida s vezanim molekulama šećera, kao i mukopolisaharidoze, u kojima se nakuplja prekomjerna količina velikog, složenog šećera molekule.
Kako se nasljeđuju lipoidoza?
Lizosomske bolesti skladištenja naslijeđuju se od jednog ili oba roditelja koji nose defektan gen koji regulira specifični enzim koji metabolizira lipide u klasi stanica u tijelu. Mogu se naslijediti na dva načina:
- Autosomno recesivno nasljeđivanje javlja se kada oba roditelja nose i prenose kopiju defektnog gena, ali nijedan roditelj nije zahvaćen poremećajem. Svako dijete rođeno od ovih roditelja ima 25 posto šanse da naslijedi obje kopije defektnog gena, 50 posto vjerojatnost da će biti nositelj sličan svojim roditeljima i 25 posto šanse da ne naslijedi nijednu kopiju neispravnog gen. Djeca oba spola mogu se naslijediti autosomno recesivno.
- X-vezano (ili spolno povezano) recesivno nasljeđivanje javlja se kada majka nosi zahvaćeni gen na X kromosomu. X i Y kromosomi sudjeluju u određivanju spola. Žene imaju dva X kromosoma, dok muškarci imaju jedan X kromosom i jedan Y kromosom. Sinovi nositeljica imaju 50 posto šanse da naslijede i utječu na poremećaj, jer sinovi dobivaju jedan X kromosom od majke i jedan Y kromosom od oca. Kćeri imaju 50 posto šanse da naslijede zahvaćeni X kromosom od svoje majke te su nositeljice ili su blago zahvaćene. Oboljeli muškarci ne prenose bolest na svoje sinove, ali će njihove kćeri biti nositeljice i nositeljice bolesti.
Vrste i simptomi lizosomskih bolesti skladištenja
Gaucherova bolest - najčešća od lizosomskih bolesti. Bolest je uzrokovana nedostatkom enzima glukocerebrozidaze, što dovodi do nakupljanja glukocerebrozida u mnogim tkivima. Masni materijal se može nakupljati u mozgu, slezeni, jetri, bubrezima, plućima i koštanoj srži.
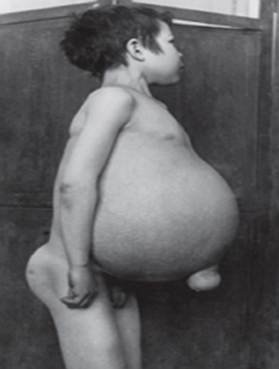
Simptomi mogu uključivati oštećenje mozga, povećanje slezene i jetra, oštećenje funkcije jetre, poremećaji skeleta i oštećenja kostiju koja mogu uzrokovati bol i prijelome, oticanje limfnih čvorova i (ponekad) susjednih zglobova, nadutost, smećkasti ton kože, anemija, nizak broj trombocita i žute mrlje na očima.
Najozbiljnije pogođeni pojedinci također mogu biti osjetljiviji na infekcije. Bolest podjednako pogađa muškarce i žene.
Gaucherova bolest ima tri opća klinička podtipa:
- Tip 1 (Ili ne neuronopatski tip) je najčešći oblik bolesti u Sjedinjenim Državama i Europi. Mozak nije zahvaćen, ali može doći do oštećenja pluća i, u rijetkim slučajevima, bubrega. Simptomi mogu početi rano u životu ili u odrasloj dobi i uključuju povećanu jetru i ozbiljno povećanje slezene, što može dovesti do rupture i dodatnih komplikacija. Slabost skeleta i bolesti kostiju mogu biti opsežne. Ljudi u ovoj skupini obično lako dobiju modrice zbog niskog broja trombocita u krvi. Također mogu osjetiti umor zbog anemije. Ovisno o nastanku i težini bolesti, osobe s tip 1 može doživjeti odraslu dob. Mnogi oboljeli imaju blagu bolest ili možda ne pokazuju nikakve simptome. Iako 1tip Gaucherova bolest je česta među ljudima aškenaskog židovskog naslijeđa i može utjecati na ljude svih etničkih pripadnosti.
- Tip 2 (ili akutna dječja neuropatija Gaucherova bolest) obično počinje unutar 3 mjeseca od rođenja. Simptomi uključuju opsežno i progresivno oštećenje mozga, spastičnost, napadaje, ukočenost udova, povećanu jetru (hepatomegalija jetre) i slezenu (splenomegalija), abnormalni pokreti očiju i slaba sposobnost sisanja i gutanja. Oboljela djeca obično umiru prije 2 godine života.
- Tip 3 (kronični neuronopatski oblik) može započeti u bilo kojem trenutku tijekom djetinjstva ili čak u odrasloj dobi. Karakteriziraju ga sporo progresivni, ali manje izraženi neurološki simptomi u usporedbi s akutnom Gaucherovom bolešću 2 vrste. Glavni simptomi uključuju poremećaje pokreta očiju, kognitivne nedostatke, lošu koordinaciju, napadaje, povećanje slezene i/ili jetre, poremećaji skeleta, poremećaji krvi uključujući anemiju i problemi s disanje. Gotovo svi s Gaucherovom bolešću 3 vrste, koji prima enzimsku nadomjesnu terapiju doći će u odraslu dob.
Za pacijente 1. i 3. vrste Enzimska nadomjesna terapija, koja se daje intravenozno svaka dva tjedna, može dramatično smanjiti veličinu jetre i slezene, smanjiti abnormalnosti skeleta i preokrenuti druge manifestacije. Uspješna transplantacija koštane srži liječi neurološke manifestacije bolesti. Međutim, ovaj postupak nosi značajne rizike i rijetko se radi u osoba s Gaucherovom bolešću.
U rijetkim slučajevima može biti potrebna operacija za uklanjanje cijele ili dijela slezene (ako osoba ima vrlo nizak broj trombocita ili kada povećani organ uvelike utječe na udobnost osobe). Transfuzija krvi može koristiti nekim osobama s anemijom. Drugi će možda trebati operaciju zamjene zgloba kako bi poboljšali mobilnost i kvalitetu života. Trenutno ne postoji učinkovit tretman za oštećenje mozga koje se može pojaviti kod ljudi s tipom 2 i 3 Gaucherove bolesti.
Niemann-Pickova bolest je skupina autosomno recesivnih poremećaja uzrokovanih nakupljanjem masti i kolesterola u stanicama jetre, slezene, koštane srži, pluća i, u nekim slučajevima, mozga.
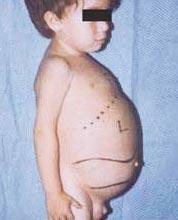
Neurološke komplikacije mogu uključivati ataksiju (nedostatak koordinacije mišića koji može utjecati na hodanje, pisanje i jelo, između ostalih funkcija), paraliza oka, degeneracija mozga, problemi s učenjem, spastičnost, otežano hranjenje i gutanje, nejasno govor, gubitak mišićnog tonusa, preosjetljivost na dodir i nešto zamućenja rožnice zbog prekomjernog nakupljanja materijala. U 50% oboljelih se oko središta mrežnice razvija karakteristična trešnja-crvena aureola, koju liječnik može vidjeti posebnim instrumentom.
Niemann-Pickova bolest je klasificirana u tri opća podtipa:
- Tip A, najteži oblik, počinje u ranom djetinjstvu. Bebe pri rođenju izgledaju normalno, ali do 6. mjeseca razvija se duboko oštećenje mozga, povećana jetra i slezena, te natečeni limfni čvorovi i čvorovi ispod kože (ksantomi). Slezena može postati 10 puta veća od svoje normalne veličine i može puknuti, uzrokujući krvarenje. Ova djeca progresivno postaju slabija, gube motoričku funkciju, mogu postati anemična i sklona su ponovnim infekcijama. Rijetko žive duže od 18 mjeseci. Ovaj oblik bolesti je najčešći u židovskim obiteljima.
- Tip B(ili juvenilni početak) obično ne utječe na mozak, ali većina djece se razvija ataksija, oštećenje živaca koji izlaze iz leđne moždine (periferna neuropatija), te plućne poteškoće koje napreduju s godinama. Povećanje jetre i slezene tipično je za adolescente. Ljudi sa tip B mogu živjeti relativno dugo, ali mnogi zahtijevaju doživotnu terapiju kisikom zbog oštećenja pluća. Niemann-Pick tipovi A i B rezultat su nakupljanja masne tvari zvane sfingomijelin zbog nedostatka enzima zvanog sfingomijelinaza.
- Tip C mogu se pojaviti rano u životu ili razviti tijekom adolescencije ili čak odrasle dobi. Niemann-Pickova bolesttip C nije uzrokovan nedostatkom sflingomijelinaze, već nedostatkom NPC1 ili NPC2 proteina. Zbog toga se razni lipidi, a posebno kolesterol nakupljaju u živčanim stanicama i uzrokuju njihov kvar. Zahvaćenost mozga može biti opsežna, što rezultira nemogućnošću gledanja gore-dolje, poteškoćama u hodanju i gutanju, progresivnim gubitkom sluha i progresivnim demencija. Ljudi sa tip C imaju samo blago povećanje slezene i jetre. Očekivano trajanje života osoba sa tip C znatno varira. Neki ljudi umiru u djetinjstvu, dok drugi koji su manje pogođeni mogu također živjeti u odrasloj dobi.
Trenutno ne postoji lijek za Niemann-Pickovu bolest. Liječenje je potporno. Djeca obično umiru od infekcije ili progresivnog neurološkog gubitka. Zabilježeni su slučajevi transplantacije koštane srži u nekoliko bolesnika s tip B s mješovitim rezultatima.
Fabryjeva bolesttakođer poznat kao nedostatak alfa-galaktozidaze-A, uzrokuje nakupljanje masne tvari u autonomnom živčanom sustavu (onom dijelu živčanog sustava koji kontrolira nevoljne funkcije kao što su disanje i rad srca), u očima, bubrezima i kardiovaskularnim sustav.
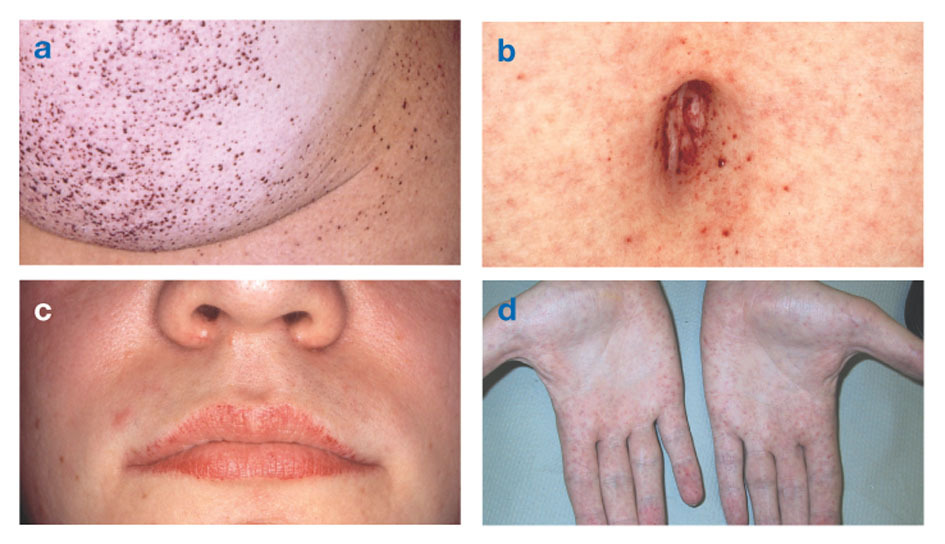
Fabryjeva bolest jedina je bolest lipida vezana uz X. Bolest uglavnom pogađa muškarce, iako se u žena javlja blaži i varijabilniji oblik. Rijetko, oboljele žene imaju teške simptome slične onima u muškaraca s ovim poremećajem. Početak simptoma obično se javlja tijekom djetinjstva ili adolescencije.
Neurološki znakovi uključuju pekuću bol u rukama i nogama koja se pogoršava na vrućem vremenu ili poslije vježba, kao i nakupljanje viška materijala u prozirnim slojevima rožnice (što dovodi do zamućenja, ali bez promjene vida). Nakupljanje masti u stijenkama krvnih žila može poremetiti cirkulaciju, dovodeći osobu u opasnost moždani udar ili srčani udar. Ostali simptomi uključuju prošireno srce, progresivno zatajenje bubregašto dovodi do završne faze bubrežne bolesti, gastrointestinalnih problema, smanjenog znojenja i groznice.
Angiokeratomi (male, nekancerozne, crvenkasto-ljubičaste podignute mrlje na koži) mogu se razviti u donjem dijelu torza i postati češći s godinama.
Osobe s Fabryjevom bolešću često prerano umiru od komplikacija zastoj srca, zatajenje bubrega ili moždani udar. Lijekovi poput fenitoina i karbamazepina često se propisuju za liječenje boli koja prati Fabryjevu bolest, ali ne i za njezino liječenje. Metoklopramid ili Lipisorb (dodatak prehrani) mogu ublažiti gastrointestinalne smetnje često se javlja kod osoba s Fabryjevom bolešću, a nekim ljudima može biti potrebna transplantacija bubrega ili dijaliza. Zamjena enzima može smanjiti skladištenje, ublažiti bol i očuvati funkciju organa kod nekih ljudi s Fabryjevom bolešću.
Farberova bolest također poznat kao Farberova lipogranulomatoza, opisuje skupinu rijetkih autosomno recesivnih poremećaja koji uzrokuju nakupljanje masne tvari u zglobovima, tkivima i središnjem živčanom sustavu. Pogađa i muškarce i žene. Bolest obično počinje u ranom djetinjstvu, ali se može pojaviti i kasnije u životu.
Razvijaju se djeca s klasičnom Farberovom bolešću neurološki simptomi, koji mogu uključivati povećanu pospanost i letargiju, te probleme s gutanje. Također mogu biti zahvaćeni jetra, srce i bubrezi. Ostali simptomi mogu uključivati kontrakture zglobova (kronično skraćivanje mišića ili tetiva oko zglobova), povraćanje, artritis, povećanje limfni čvorovi, upala zglobova, promuklost i kvržice ispod kože koje se zadebljavaju oko zglobova kako napreduje bolesti.
Pogođenim osobama s poteškoćama s disanjem možda će trebati umetnuti cijev za disanje. Većina djece s ovim stanjem umire do 2 godine, obično od plućne bolesti. U jednom od najtežih oblika bolesti, povećana jetra i slezena mogu se dijagnosticirati ubrzo nakon rođenja. Bebe rođene s ovim oblikom bolesti obično umiru unutar 6 mjeseci.
Farberova bolest je uzrokovana nedostatkom enzima zvanog ceramidaza. Trenutno ne postoji specifičan tretman za Farberovu bolest. Za ublažavanje boli mogu se propisati kortikosteroidi. Transplantacija koštane srži može smanjiti granulome (male mase upaljenog tkiva) u osoba s blagom upalom ili u bolesnika bez komplikacija na plućnom ili živčanom sustavu. U starijih osoba granulomi se mogu smanjiti ili kirurški ukloniti.
Gangliozidoza sastoji se od dvije različite skupine genetskih bolesti. Oba su autosomno recesivna i podjednako utječu na muškarce i žene.
GM1 gangliozidoza.
GM1 gangliozidoza je uzrokovana nedostatkom enzima beta-galaktozidaze, što rezultira abnormalnim nakupljanjem kiseli lipidni materijali, posebno u živčanim stanicama središnjeg i perifernog živčanog sustava sustava. GM1 gangliozidoza ima tri kliničke manifestacije:
- GM1 (najteži podtip, javlja se ubrzo nakon rođenja) može uključivati neurodegeneraciju, napadaje, povećanje jetre i slezene, grubost crta lica, nepravilnosti skeleta, ukočenost zglobova, nadutost, slabost mišića, pretjerana reakcija na prepad i problemi s hod. Otprilike polovica oboljelih razvije mrlje boje trešnje u oku. Djeca mogu biti gluha i slijepa do 1. godine i često umrijeti do 3. godine od srčanih komplikacija ili upala pluća.
- Kasno dijete GM1 gangliozidoza obično počinje u dobi od 1 do 3 godine. Neurološki simptomi uključuju ataksiju, napadaje, demenciju i poteškoće u govoru.
- GM1 gangliozidoza razvija se u dobi od 3 do 30 godina. Simptomi uključuju smanjenu mišićnu masu (atrofiju mišića), neurološke komplikacije koje su manje teške i napreduju sporije od drugih oblika poremećaji, zamućenje rožnice kod nekih ljudi i trajne kontrakcije mišića koje uzrokuju uvijanje i ponavljajuće pokrete ili abnormalne položaje (distonija). Angiokeratomi se mogu razviti u donjem dijelu tijela. Veličina jetre i slezene je normalna kod većine oboljelih osoba.
GM2 gangliozidoza.
GM2 gangliozidoza također uzrokuje da tijelo nakuplja višak kiselih masnih tvari u tkivima i stanicama, prvenstveno u živčanim stanicama. Ovi poremećaji nastaju zbog nedostatka enzima beta-heksosaminidaze. GM2 poremećaji uključuju:
- Tay-Sachsova bolest (također poznat kao GM2 gangliozidoza varijanta B) i njezini varijanti oblici uzrokovani su nedostatkom enzima heksosaminidaze A. Oboljela djeca se normalno razvijaju tijekom prvih nekoliko mjeseci života. Simptomi počinju u dobi od 6 mjeseci i uključuju progresivni gubitak mentalnih sposobnosti, demenciju, smanjen kontakt očima, povećan zapanjujući odgovor na buku, progresivni gubitak sluha koji dovodi do gluhoće, otežano gutanje, sljepoća, trešnje crvene mrlje na mrežnici i neke paraliza. Napadaji mogu početi u drugoj godini djetetova života. Bebe s vremenom mogu postati ovisne o sondi za hranjenje i često umrijeti u dobi od 4 godine od ponavljajućih infekcija. Nažalost, ne postoji poseban tretman. Antikonvulzivi u početku mogu kontrolirati napadaje. Ostali potporni tretmani uključuju pravilnu prehranu i hidrataciju te metode za održavanje dišnih putova otvorenim. Rijedak oblik poremećaja tzv kasna Tay-Sachsova bolest, javlja se u osoba u dobi od 20 do 30 godina i karakterizira ga nestabilan hod i progresivno neurološko pogoršanje.
- Sandhoffova bolest (Opcija AB) je teški oblik Tay-Sachsove bolesti. Početak se obično javlja u dobi od 6 mjeseci i nije ograničen ni na jednu etničku skupinu. Neurološki znakovi mogu uključivati progresivno pogoršanje središnjeg živčanog sustava, motor slabost, rano sljepilo, teška reakcija uplašenosti na zvuk, spastičnost, šok ili trzanje mišići (mioklonus), epilepsija, abnormalno povećana glava (makrocefalija) i crvene mrlje u oku. Ostali simptomi mogu uključivati česte respiratorne infekcije, šum srca, crte lica poput lutke i povećanje jetre i slezene. Ne postoji specifičan tretman za Sandhoffovu bolest. Kao i kod Tay-Sachsove bolesti, potporna njega uključuje održavanje dišnih putova otvorenim u svakom trenutku, kao i pravilnu prehranu i hidrataciju. Antikonvulzivi u početku mogu kontrolirati epilepsiju (napadaje).
Krabbeova bolest(također poznat kao leukodistrofija globularnih stanica i galaktozilceramidna lipidoza) je autosomno recesivna bolest uzrokovana nedostatkom enzima galaktocerebrozidaze. Bolest najčešće pogađa dojenčad s početkom prije 6 mjeseci starosti, ali se može pojaviti tijekom adolescencije ili odrasle dobi.
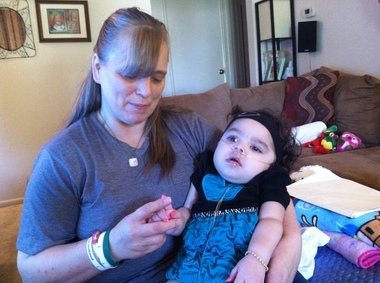
Nakupljanje neprobavljenih masti utječe na rast zaštitne ovojnice živaca (mijelinske ovojnice) i uzrokuje ozbiljno oštećenje mentalnih i motoričkih sposobnosti. Ostali simptomi uključuju slabost mišića, smanjenu sposobnost mišića istezanja (hipotonija), napetost mišića (spastičnost), iznenadni šok ili trzanje udova (mioklonični napadaji), razdražljivost, neobjašnjiva groznica, gluhoća, sljepoća, paraliza i poteškoće pri gutanju. Može doći i do dugotrajnog gubitka težine.
Bolest se može dijagnosticirati ispitivanjem enzima i utvrđivanjem njezinih karakterističnih skupina stanica u globoidna tijela u bijeloj tvari mozga, demijelinizaciju živaca i degeneraciju te uništavanje moždanih stanica. U dojenčadi bolest je obično smrtonosna prije 2 godine života. Osobe s uznapredovalim oblikom bolesti imaju blaži tijek bolesti i žive znatno dulje.
Nije razvijeno specifično liječenje za Krabbeovu bolest, iako rana transplantacija koštane srži može pomoći nekim ljudima. Osobe s uznapredovalim oblikom bolesti imaju blaži tijek bolesti i žive znatno dulje.
Metakromatska leukodistrofija, ili MLD, skupina je poremećaja obilježenih nakupljanjem masnih tvari u bijeloj tvari središnjeg živčanog sustava te u perifernim živcima i donekle u bubrezima. Slično Krabbeovoj bolesti, MLD utječe na mijelin, koji pokriva i štiti živce. Ovaj autosomno recesivni poremećaj uzrokovan je nedostatkom enzima arilsulfataze A. Ovaj poremećaj pogađa i muškarce i žene.
Metakromatska leukodistrofija ima tri karakteristična oblika: kasno dojenčad, juvenilni i odrasli.
- Kasno dojenčad MLD obično počinje između 12 i 20 mjeseci nakon rođenja. Djeca se isprva mogu činiti normalnim, ali imaju poteškoće u hodu i sklonost padu, praćeni povremenim bolovima u rukama i nogama. progresivni gubitak vida koji dovodi do sljepoće, kašnjenja u razvoju i gubitka prethodno stečenih prekretnica, otežanog gutanja, napadaja i demencije do 2 godine. Djeca također razvijaju progresivno trošenje mišića i slabost te na kraju gube sposobnost hodanja. Većina djece s ovim oblikom poremećaja umire do 5. godine.
- Maloljetnički MLD obično počinje u dobi od 3 do 10 godina. Simptomi uključuju narušen školski uspjeh, mentalno oštećenje, ataksiju, napadaje i demenciju. Simptomi napreduju, a smrt nastupa 10-20 godina nakon pojave bolesti.
- Forma za odrasle. Simptomi odrasle osobe počinje nakon 16. godine i može uključivati ataksiju, napadaje, abnormalni tremor udova (drhtanje), smanjenu koncentraciju, depresija, mentalnih poremećaja i demencije. Smrt obično nastupa 6-14 godina nakon pojave simptoma.
MLD se ne može izliječiti. Liječenje je simptomatsko i potporno. Transplantacija koštane srži u nekim slučajevima može odgoditi napredovanje bolesti. Značajan napredak postignut je s genskom terapijom na životinjskim modelima s MLD i u kliničkim ispitivanjima.
Wolmanova bolesttakođer poznat kao nedostatak lizosomske kisele lipazeje ozbiljan poremećaj skladištenja lipida koji je obično fatalan u dobi od 1 godine. Ovaj autosomno recesivni poremećaj karakterizira nakupljanje estera kolesterola (obično transportni oblik kolesterola) i trigliceridi (kemijski oblik u kojem masti postoje u tijelu), koji se mogu značajno akumulirati i uzrokovati oštećenje stanica i tkanine.
Od ovog poremećaja pate i muškarci i žene. Dojenčad je normalna i aktivna pri rođenju, ali brzo razvija progresivno mentalno oštećenje, povećanu jetru i jako povećanu slezinu, nadutost, gastrointestinalne probleme. žutica, anemija, povraćanje i naslage kalcija u nadbubrežne žlijezdeuzrokujući njihovo stvrdnjavanje.
Druga vrsta nedostatak lizosomske kisele lipaze je bolest skladištenja estera kolesterola. Ova iznimno rijetka bolest nastaje kao posljedica skladištenja estera kolesterola i triglicerida u krvnim stanicama, limfa i limfoidno tkivo. U djece u odrasloj dobi povećava se jetra, što dovodi do ciroze i kronične zatajenje jetre. Djeca mogu imati i naslage kalcija u nadbubrežnim žlijezdama, a žutica se može razviti u kasnijim fazama bolesti.
Trenutno se pitanje zamjene enzima aktivno proučava i kod Wolmanove bolesti i kod nakupljanja estera kolesterola.
Pažnja! Informacije su samo u informativne svrhe i nisu namijenjene postavljanju dijagnoze i propisivanju liječenja. Uvijek se posavjetujte sa svojim liječnikom specijalistom!


