Laron -szindróma: mi ez, okok, tünetek, kezelés, prognózis
Tartalom
- Mi a Laron -szindróma?
- Történelem
- jelek és tünetek
- Okoz
- Érintett populációk
- Tüneti rendellenességek
- Diagnosztika
- Standard kezelések
- Előrejelzés
Mi a Laron -szindróma?
Laron -szindróma Rendkívül ritka genetikai rendellenességek csoportja, amelyekben a szervezet nem tudja használni a növekedési hormont (rövid. GR). A Laron -szindrómát a növekedési hormon receptor (GH) génjének mutációi vagy a gének mutációi okozhatják részt vesz a sejt működési útvonalában, miután a növekedési hormon kötődik a receptorához, megakadályozva a termelődését inzulinszerű növekedési faktor 1 (IGF-1, IGF-1), a növekedési hormon szervezetre gyakorolt hatásáért felelős anyag. Még ritkábban, a GH gén deléciójában szenvedő, rekombináns GH -val kezelt gyermekeknél antitestek alakulnak ki, amelyek gátolják a növekedési hormon receptorhoz való kötődését. Az érintett gyermekek nem tudnak normálisan növekedni.
Azok a gyermekek, akiknek STH-hiánya van, és akiket a pubertás előtt IGF-1-gyel kezelnek, javult a növekedésük, de A rekombináns GH -kezelésben részesülő GH -hiányos gyermekekkel ellentétben nem térnek vissza a normális állapotból magasság. Ezen állapotok kezelése csak akkor hatékony, ha a növekvő csontok még nyitva vannak, azaz a serdülőkor vége előtt. Az IGF-1 érzéketlensége az IGF-1 receptor mutációja miatt utánozza a Laron-szindrómát, de kevésbé súlyos növekedési kudarcot eredményez, és némileg reagál a rekombináns GH kezelésre.
A Laron -szindrómát a csontok rövid és lassú növekedése, valamint a normál és a magas keringő növekedési hormon szint jellemzi. További gyakori tünetek a késleltetett pubertás, kidülledő homlok, alacsony vércukorszint csecsemőkorban és kisgyermekkorban, és elhízott felnőttkorban. Kivéve a szindróma rendkívül ritka formáját, ahol az IGF-1 gén hibás, de az agy fejlődése normális, nyilvánvalóan azért, mert az IGF-1 a magzat élete során nyerhető anélkül, hogy másokban növekedési hormont stimulálna körülmények. Néhány, de határozottan nem minden olyan beteg, akinek ritkábban fordul elő IGF-1 receptor hiánya, enyhe értelmi fogyatékos lehet.
Történelem
Laron és izraeli kollégái először 1966 -ban számoltak be az STH hiányáról, az 1958 -ban kezdődött és a mai napig tartó megfigyelések alapján. Az általuk leírt szindróma molekuláris alapja - néhány izraeli betegnél először történt az STH gén mutációja 1989 -ben írták le, és azóta sok kutató több mint 60 különböző mutációt talált a génben erre mókus. Az elmúlt 15 évben leírtak olyan génmutációkat, amelyek a növekedési hormon hatásának útjában vannak a növekedési hormonhoz való kötődésük után, és amelyek az IGF-1 hiány különböző hatásaihoz kapcsolódnak.
jelek és tünetek

A tünetek széles skálája létezik az érintett génmutációktól függően (lásd alább). szakasz "Okok"). Nagyon kevés IGF-1 génmutációval rendelkező ember rendelkezik súlyos értelmi fogyatékossággal és méhen belüli növekedési zavarokkal, süketséggel és mikrognátiával. A GH hiány súlyos növekedési kudarcot eredményez anélkül, hogy hátrányosan befolyásolná a méhen belüli növekedést vagy az agy fejlődését, valamint a mutációt STAT5b, felelős egy fontos aktiváló fehérjeért, hasonló hatással van a növekedésre, de az immunkompetencia súlyos károsodásával is jár. IGFALS A keringő IGF-1 fontos stabilizáló alkotórészét érintő mutációk, bár nagyon alacsony keringő IGF-szintekhez kapcsolódnak, csak mérsékelt hatással vannak a növekedésre.
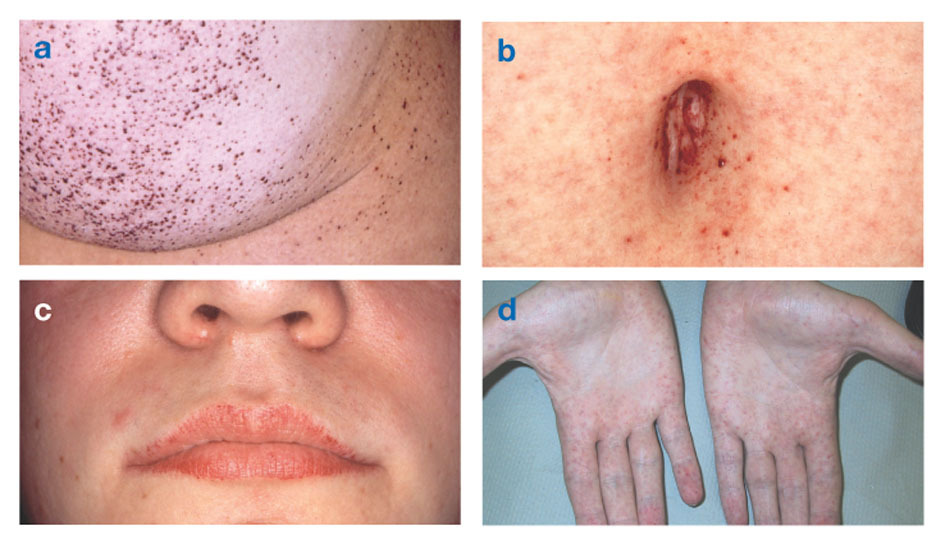
A Laron -szindrómát súlyos, de arányos alacsony termet jellemzi, amely a születéskor kezdődő növekedési rendellenességből ered. A növekedés késleltetésével együtt a fogzás is késik. Aránytalanság van a koponya és az arc növekedése, a nyereg orra és a mélyen fekvő szemek között is. A szexuális fejlődés mindkét nemnél mérsékelten késik. Az ilyen rendellenességekben szenvedő nőknél a menstruáció általában 16-19 éves kor között jelentkezik. A karok és a lábak a szokásosnál kisebbek a teljes testmérethez képest. Továbbá, felnőtt betegeknél magas hang és elhízottságfőleg nőknél.
Gyermekeknél magas a GH vérszintje, de felnőttek stimulálása nélkül nem nyilvánvaló. A fiatal betegek nagy százalékának alacsony a vércukorszintje (hipoglikémia), ami egyes nagyon kisgyermekek rohamaival járhat. A közelmúltban a kutatók azt találták, hogy a GH-hiányos populációban Ecuadorban (ahol a világ lakosságának körülbelül 1/3-át azonosították a GH-receptorok hiányával) nem volt rák és cukorbetegség molekuláris bizonyítékokkal a DNS -öregedés elleni változások elleni védelemről. Ennek oka lehet az IGF-1 alacsony szintje elleni védő hatás, és az elhízás ellenére a cukorbetegség hiányában a növekedési hormon ellenszabályozó hatásainak hiánya miatt.
Okoz
A Laron -szindróma autoszomális recesszív genetikai rendellenességként öröklődik, és a növekedési hormon gén mutációja okozza vagy a sejt hatásmechanizmusában részt vevő gének mutációi után, miután a GH a receptorhoz kötődik, beleértve STAT5b, IGF-1 és IGFALS.
A recesszív genetikai rendellenességek akkor fordulnak elő, amikor egy személy örököl két kópiát egy azonos tulajdonságú kóros génből, egyet minden szülőtől. Ha egy személy egy normális gént és egy gént örököl a betegségre, akkor ő lesz a betegség hordozója, de általában tünetmentes. A kockázat két hordozó szülő esetében, akik mindketten továbbítják a megváltozott gént, és megfertőzik a babát, minden terhesség esetén 25%. Minden gyermek terhességével 50% annak kockázata, hogy gyermeke lesz, aki hordozó lesz, mint a szülő. 25%annak a valószínűsége, hogy a gyermek normális géneket kap mindkét szülőjétől. A kockázat férfiaknál és nőknél azonos.
Azok a szülők, akik közeli rokonok (testvér), nagyobb kockázatnak vannak kitéve, mint a nem rokonok olyan szülők, akiknek ugyanaz a kóros génjük van, ami növeli a recesszív genetikájú gyermek születésének kockázatát rendellenesség.
Érintett populációk
Világszerte csak körülbelül 300 Laron -szindróma esetet jelentettek STH -hiány miatt. A bejelentett esetek többségének (90%) etnikai hovatartozása ismert. A világszerte azonosított szindrómás, körülbelül 250 érintett személy körülbelül kétharmada sémi, a többi fele mediterrán vagy dél-ázsiai származású. A sémi csoport magában foglalja az arab, keleti vagy közel -keleti zsidó lakosságot és a legnagyobb csoportot, genetikailag homogén 100+ Converse Ecuadorban (zsidók, akik közben kereszténységre tértek Inkvizíció).
Tüneti rendellenességek
Az alábbi betegségek tünetei hasonlóak lehetnek a Laron -szindrómához. Az összehasonlítások hasznosak lehetnek a differenciáldiagnosztika során:
A Laron -szindróma genetikai formái mellett a megfelelő GH -termelés ellenére a normális növekedés képtelensége számos betegségben gyakori, beleértve a krónikus betegségeket, az alultápláltságot, vesebetegség, májbetegség és veleszületett szindrómák.
- Koporsó-Siris szindróma Ismeretlen eredetű betegség. A születéskor jelen van, és mindkét nemet érinti. A betegséget elsősorban táplálkozási problémák, gyakori légúti fertőzések és gyenge növekedés jellemzi.
- Cockayne -szindróma Progresszív rendellenesség, amely az élet második évében nyilvánul meg. A napfényre való fokozott érzékenység és a növekedés visszamaradása jellemzi.
- Növekedési hormon hiány (NDK) legsúlyosabb formájában nagyon hasonlít az elsődleges Laron -szindrómához, a fő különbség az a Laron -szindrómás gyermekek magas GH vérszinttel rendelkeznek, és a rekombináns GH beadása nem vezet normalizációhoz növekedés. A GHR oka gyakran ismeretlen, de a növekedési hormon receptor felszabaduló genetikai hibái az agyból és a GH -molekulából származó hormon, valamint olyan tényezők, amelyek a hormon mellett más hormonok hiányához vezetnek növekedés. Meglehetősen könnyen és megbízhatóan kezelhető rekombináns GH injekciókkal.
- Hydrocephalus - az állapot összetéveszthető a Laron -szindrómával a csecsemőkori STH -hiány miatt a homlok domborulata, jelenléte miatt a szemhártya (tunica albuginea) a szem írisze felett, a koponyához képest kicsi arc és a domború erek fej. Laron -szindrómában azonban nem látható gyors fejnövekedés, amint ez a hydrocephalus esetében is megfigyelhető.
Sokkal több rendellenességet okozhat a rövid termet.
Diagnosztika
A Laron -szindróma diagnózisa általában akkor történik, amikor a növekedési képtelenséget egy tipikus betegség kíséri az arc megjelenése és a központi puffadás, ami GH hiányra utal, de a megnövekedett szint azonosításával GR.
Standard kezelések
A ritka betegségben szenvedő rhIGF-1 gyógyszert (rekombináns IGF-1) olyan gyermekek számára engedélyezték, akiknél a növekedési zavar GH-hiányhoz vagy GH-inaktiváló antitestekhez kapcsolódik.
A Laron -szindróma kezelése rekombináns humán GH -val nem hatékony, mert a szervezet nem tudja használni a hormont a növekedéshez. A rekombináns IGF-1 terápia a hipoglikémia kockázatával jár, amelyet etetéssel meg lehet előzni. Szerencsére a hipoglikémia hosszú távú hatásait nem vették észre.
A betegeknek és családtagjaiknak is ajánlott genetikai tanácsadás.
Előrejelzés
A hosszú távú prognózis normálisnak tűnik az STH-hiányban és a receptor utáni hibákban szenvedő betegeknél, a mutáció kivételével STAT5b. Valójában az STH -hiányos alanyok, annak ellenére, hogy elhízottak, fokozott érzékenységet mutatnak inzulin és nem alakul ki cukorbetegségük, és védhetők a rák ellen is.
Mutációval rendelkező emberek STAT5bvalószínűleg alacsony a várható élettartam a krónikus betegség következtében tüdőbetegségek.