Onvolmaakte osteogenese
 Onvolmaakte osteogenese is een aangeboren ziekte van botten, evenals individuele bindweefselstructuren. Een andere naam voor onvolmaakte osteogenese is een ziekte van kwetsbare botten. De ziekte wordt door erfenis overgedragen, d.w.z. Is genetisch bepaald.
Onvolmaakte osteogenese is een aangeboren ziekte van botten, evenals individuele bindweefselstructuren. Een andere naam voor onvolmaakte osteogenese is een ziekte van kwetsbare botten. De ziekte wordt door erfenis overgedragen, d.w.z. Is genetisch bepaald.
Onvolledige osteogenese optreedt bij zowel mannen als vrouwen, de incidentiepercentage is 1 pasgeboren per 12 000-15 000 kinderen. Deze pathologie werd voor het eerst genoemd in de 17e eeuw. Bij onvolmaakte osteogenese worden groei, huidletsels, spierweefsel, bloedvaten, tanden, pezen en gehoororganen gestoord.
Ondanks de moeilijkheden waarmee mensen met onvolmaakte osteogenese geconfronteerd worden, leiden de meeste patiënten een volwaardige levensstijl. Op dit moment wordt de behandeling van onvolmaakte osteogenese steeds populairder. Niettemin is het nog steeds onmogelijk om een patiënt volledig te genezen met zulke ziekte.
Onvolmaakte osteogenese van de oorzaak van
Onvolmaakte osteogenese verwijst naar heterogene erfelijke ziekten die het bindweefsel beïnvloeden. De ziekte wordt beschreven door osteopenie en enkele andere klinische tekenen van osteogenese.
De belangrijkste oorzaak van deze pathologische ontwikkeling is een erfelijke mutatie van collageengenen. Zeldzaam is het spontane voorkomen van mutaties in de genen van dit eiwit. Het gevolg van genmutatie is een overtreding van collageensynthese, vandaar de pathologieën bij de vorming van bot- en kraakbeenweefsels.
Onvoldoende hoeveelheid gesynthetiseerde collageen( niet gemuteerd) is ook een van de redenen voor de ontwikkeling van onvolmaakte osteogenese. In dit geval gaat de ziekte op in een zachtere vorm. Onvolmaakte osteogenese van deze aard wordt uitgedrukt in individuele fracturen van de ledematen, na de puberteit neemt het aantal fracturen in de regel af.
Moderne geneeskunde onderscheidt verschillende soorten onvolmaakte osteogenese, waarbij rekening wordt gehouden met radiografische, klinische, collageen proteogene veranderingen.
Onvolmaakte osteogenese types:
Type I - een zwak uitgedrukte vorm, een dominant type erfdeel. Onvolmaakte osteogenese, gekenmerkt door brose botten, de aanwezigheid van blauwe sclera.
Type II - Perinatal-dodelijk.
Type III - Deformatie van het skelet is progressief.
Type IV - het dominante type erfenis, de vervormingen van het skelet worden niet sterk uitgesproken, sclera zijn normaal.
Velen geloven dat onvolmaakte osteogenese zich ontwikkelt tegen de achtergrond van kwalitatieve en kwantitatieve schendingen van collageen type I synthese. Onvolmaakte osteogenese van type I wordt gekenmerkt door een verminderd niveau van synthese van normaal collageen. Type II, zoals type IV onvolmaakt osteogenese, wordt veroorzaakt door een afname van de totale hoeveelheid collageen door een verminderde stabiliteit, hoewel het collageensyntheseproces zonder verstoringen passeert.
Onvolmaakte osteogenese symptomen
Elk type imperfecte osteogenese heeft zijn karakteristieke symptomatologie.
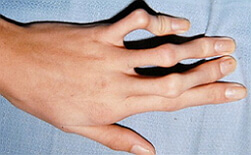
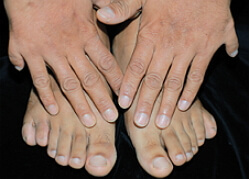
Dus, ik type onvolmaakte osteogenese bij kinderen wordt gemanifesteerd door osteoporose, evenals frequente botbreuken. Na een 10-jarige leeftijd neemt de incidentie van fracturen af, maar komt na 40 jaar weer op. De blauwe sclera en de vroege verschijning van de seniele rand worden genoteerd. Patiënten( ongeveer 50% van de gevallen) met dit type onvolmaakte osteoporose van lage groei, sommige van dentine blijft normaal, sommige patiënten hebben dentine opaal. Bij type I ontstaan aorta veranderingen in een aantal gevallen, er is een mitral defect( bij 20% van de gevallen voorkomt de mitralklep prolaps), neusbloeding.
Type II van onvolmaakte osteogenese wordt gekenmerkt door foetale dood voor de geboorte of vroege neonatale dood. Breuken zijn veelvoudig en frequent, gemakkelijk voorkomen. Er zijn 3 groepen:
Groep A. Schade aan het hoofd, ledematen in de foetus worden opgemerkt tijdens de zwangerschap door de breekbaarheid van bindweefselformaties. Het hersengebied van de schedel is vergroot, de thorax is integendeel verminderd, de ledematen zijn kort en gebogen. Er zijn ernstige gevallen van verkalking van de aorta muren en endocardium. Dergelijke kinderen zijn zeer kleine groei geboren( ongeveer 25-30 cm).
Geboren voor de periode, met 20% van dergelijke vroegtijdige geboorten - stilgeboorte. Sommige zieke kinderen sterven in de vroege dagen, sommige wonen tot de 4e week. Pathologische veranderingen kunnen worden gedetecteerd zelfs voor de geboorte van het kind met behulp van röntgenstudie: de femur is breed, er zijn golvende randen, ribben met streepjes, de thorax is kort. Bij het uitvoeren van medische genetische counseling in probanden is er een mogelijkheid van een molecuul defect gecombineerd met heterozygotheid van mutaties gelokaliseerd in het collageen gen. De erfenis is autosomaal dominant.
Groep B. Fenotypisch vergelijkbaar met groep A. Desalniettemin zijn storingen in het ademhalingsstelsel niet zo uitgesproken, kinderen met onvolmaakte osteogenese leven al meerdere jaren. Er zijn verkorte buisvormige botten en veranderingen in de structuur van de ribben, maar hun fracturen zijn zeldzaam. Vermoedelijke erfelijkheid is autosomaal recessief.
Groep B. Zulke gevallen van onvolmaakte osteogenese zijn zeldzaam. Gekenmerkt door stilgeboorte of het begin van de dood in de eerste maand. Bij patiënten, kleine groei, fijnheid van buisvormige botten( met name diafyse), worden schedelbeentjes zonder ossificatie genoteerd. Vermoedelijk een autosomaal recessief type erfgoed.
Patiënten met onvolmaakte osteogenese van type III zijn zeldzaam. Tijdens de bevalling ontstaan fracturen soms, het lichaam van de pasgeboren is verkort, terwijl de massa binnen normale grenzen mag zijn. O-vormige vervorming van vrije extremiteiten, kyphoscoliosis is kenmerkend. De oorzaak van de dood van ongeveer de helft van de patiënten is verschillende veranderingen in het skelet en het bloedsomloopstelsel. Osteoporose wordt uitgesproken, de groei van de botten in lengte is gebroken samen met hun sinificatie. In de groeizones van botweefsel wordt ongewenste verkalking waargenomen, wat leidt tot het ontstaan van spotting( de zogenaamde "maïskernen").Erfelijkheid is niet precies bekend.
IV type onvolmaakte osteogenese bij kinderen is meestal gemanifesteerd als een stoornis in het skelet. Dit type heeft een grote variabiliteit van osteopenie, het aantal botfracturen, leeftijd, blueness sclera. Gewoonlijk met leeftijd neemt het aantal botfracturen af, botroussen ontstaan na 30 jaar bij sommige patiënten met een onvolmaakte osteogenese van dit type, het gehoor wordt verstoord. Volgens de toestand van dentin zijn patiënten verdeeld in 2 groepen: sommige hebben scherpe veranderingen in dentine( opale tanden), anderen hebben geen veranderingen in dentine.
Onvolmaakte osteogenese behandeling
Diagnose van onvolmaakte osteogenese berust in de eerste plaats op het klinische beeld van de ziekte. Om een nauwkeurige diagnose vast te stellen, is een specialistisch overleg( endocrinoloog, orthopedist of geneticus) en de aanstelling van laboratoriumstudies nodig om andere ziekten uit te sluiten.
Laboratoriummethoden omvatten moleculaire analyse van collageen en zijn biochemische analyse, onderzoek van huidbiopsiemateriaal. Instrumentale methoden van de studie omvatten: röntgenonderzoek( bepaling van osteopenie, verschillende fracturen, buigen van botten, fracturen van vertebrale kolommen, intercalaire botten in de schedel), botbiopsie, densitometrie.
Het hoofddoel van de behandeling van onvolmaakte osteogenese is het verminderen van het aantal fracturen in de patiënt, verhoging van de mineralisatie van botten om hun breekbaarheid te verminderen, de botmassa te verhogen. Een belangrijk punt van behandeling is de aanpassing van het kind, alsook van zijn ouders, aan de veranderde levensstijl door ziekte. Dit voorkomt in de toekomst fracturen van botten.
De volledige behandeling bestaat uit:
- Medicatie. Om onvolmaakte osteogenese te bestrijden worden preparaten gebruikt: bisfosfonaten, groeihormoon, vitamine D3, calciumgluconaat, magnesiumzouten en kaliumzouten, ergocalciferol, complexonen, glycerofosfaat.
- Operatieve interventie. Het is nodig om fracturen te elimineren, botten te versterken, correcte veranderingen te wijten aan vervorming, prothese. Soms is de chirurgische behandeling niet één operatie: vanwege de groei van de botten hebben eerder ingebouwde pennen vervang nodig.
Bij de behandeling van fracturen worden flexibele titaniumstaafjes met hypoallergenische eigenschappen gebruikt. Dergelijke staven zijn goed vastgemaakt met een fractuur, zodat de behoefte aan extra immobilisatie van het lichaamsgebied verdwijnt. Het gebruikelijke gebruik van gipsverbindingen zorgt voor problemen in de zorg, het veroorzaakt nieuwe fracturen. Het is ook mogelijk om verschillende graften, botplastica en andere metalen structuren te gebruiken.
Gezien de toegenomen breekbaarheid van botten in een patiënt, in sommige gevallen, orthopedische producten osteoclasis de vervorming van bot wijzigingen en het been vast is gips corrigeren. Ook geschikt voor het immobiliseren van lichaamsrekken.
aanvulling op de basis behandelingswerkwijzen van osteogenesis imperfecta worden ook gebruikt: oefenen, fysiotherapie, osteosynthese. Voor zowel de patiënt als de familieleden wordt een opleiding van rehabilitatie en psychotherapeutische ondersteuning verstrekt.
volledige verloop van de behandeling niet volledig gaan met zulke ernstige ziekte zoals osteogenesis imperfecta, zodat al het bovenstaande methoden is het mogelijk slechts ten dele van de symptomen te elimineren en het gemak van de manier van leven voor de patiënt zonder dat de onderliggende oorzaak van de ziekte.