Zespół Waardenburga: co to jest, objawy, leczenie, rokowanie
Treść
- Co to jest zespół Waardenburga?
- Symptomy i objawy
- Powoduje
- Rodzaj dziedziczenia
- Dotknięte populacje
- Zaburzenia objawowe
- Diagnostyka
- Zabiegi standardowe
- Prognoza
Co to jest zespół Waardenburga?
Zespół Waardenburga Jest zaburzeniem genetycznym, które może ujawnić się przy urodzeniu (wrodzone). Zakres i nasilenie powiązanych objawów i oznak może się znacznie różnić w zależności od przypadku. Jednak pierwotne objawy często obejmują charakterystyczne nieprawidłowości twarzy; niezwykle zmniejszone zabarwienie (pigmentacja) włosów, skóry i/lub tęczówki obu oczu; i/lub wrodzona głuchota. Mówiąc dokładniej, niektórzy pacjenci mogą mieć niezwykle szeroki grzbiet nosa z powodu bocznego (bocznego) przemieszczenia wewnętrznych kącików oczu (telekant). Ponadto nieprawidłowości pigmentacyjne mogą obejmować białe pasmo włosów rosnące nad czołem; przedwczesne siwienie lub wybielanie włosów; różnice w kolorze dwóch tęczówek lub w różnych częściach tej samej tęczówki (heterochromia tęczówki); i (lub) nierówne, nienormalnie jasne (odbarwione) obszary skóry (leukoderma). Niektóre osoby mogą również mieć upośledzenie słuchu z powodu nieprawidłowości w uchu wewnętrznym (ubytek słuchu czuciowo-nerwowego).
Badacze opisali różne typy zespołu Waardenburga (dalej skrót. SV) na podstawie powiązanych objawów i określonych danych genetycznych. Na przykład zespół Waardenburga typu I (CB1) jest zwykle związany z bocznym przemieszczeniem wewnętrznego kącika oka (tj. teleskopowy), ale typ II (CB2) nie jest związany z tą cechą. Ponadto wiadomo, że CB1 i CB2 są spowodowane zmianami (mutacjami) w różnych genach. Inna postać, znana jako typ III (CB3), w której charakterystyczne rysy twarzy, oka (okularu) i zaburzenia słuchu mogą być związane z charakterystycznymi wadami rozwojowymi ramion i dłoni (kończyny górne). Czwarta postać, znana jako CB4 lub choroba Waardenburga-Hirschsprunga, może wykazywać pierwotne objawy SV w połączeniu z Choroba Hirschsprunga. To ostatnie to zaburzenie przewodu pokarmowego (jelitowego), w którym nie ma grupy wyspecjalizowanych ciał komórek nerwowych w gładkiej (mimowolnej) ścianie mięśniowej grubej jelita.
W większości przypadków zespół Waardenburga jest przenoszony w sposób autosomalny dominujący. Zidentyfikowano szereg różnych genów chorobowych, które mogą powodować zespół Waardenburga u niektórych osób lub rodzin (krewnych).
Symptomy i objawy
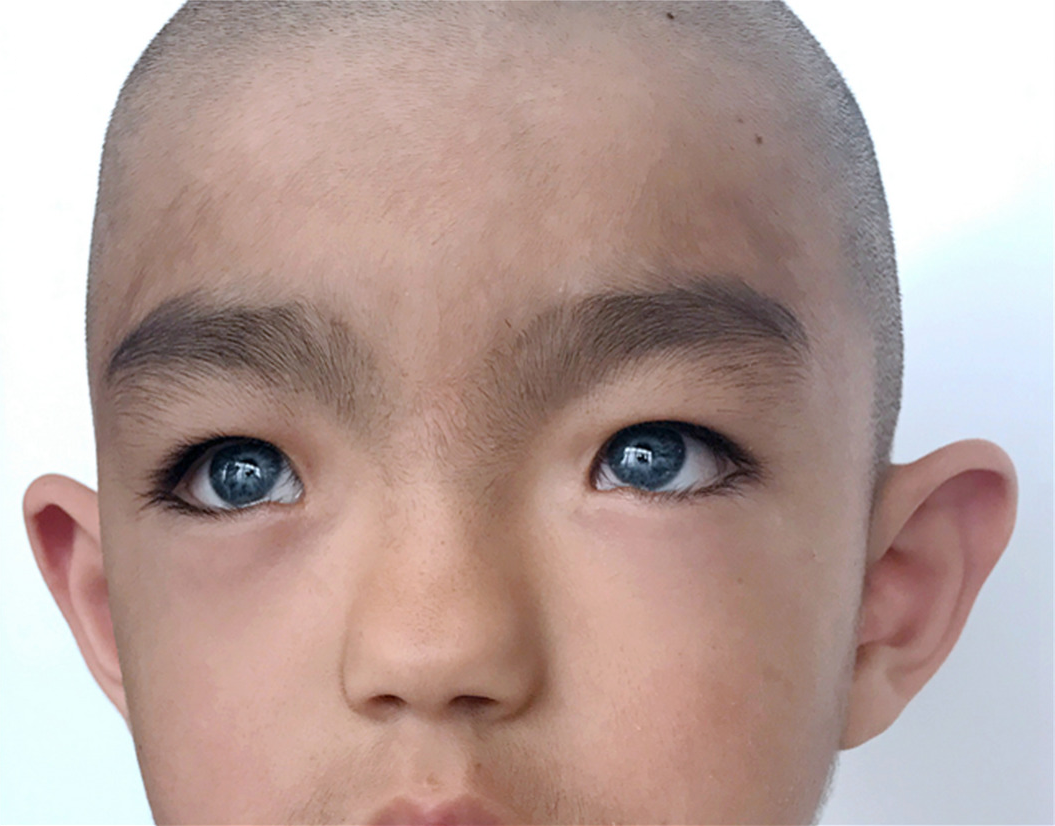
Podstawowe objawy zespołu Waardenburga mogą obejmować:
- charakterystyczne anomalie twarzy (patrz. Zdjęcie);
- zaburzenia pigmentacji (hipopigmentacja) włosów, skóry i/lub tęczówki lub tęczówki obu oczu (częściowe bielactwo);
- wrodzona utrata słuchu.
Jednak, jak wspomniano wcześniej, związane z tym objawy i oznaki mogą być bardzo różne, w tym wśród dotkniętych chorobą członków rodziny. Na przykład, podczas gdy niektórzy pacjenci mogą mieć tylko jedną cechę, inni mogą mieć wiele nieprawidłowości związanych z chorobą.
U niektórych pacjentów z zespołem Waardenburga (SV) dochodzi do nieprawidłowego przemieszczenia bocznych (bocznych) wewnętrznych kącików oczu, utworzonych przez połączenie górnej i dolnej powieki (telekant). Ponadto stan ten może być związany z niezwykle małymi otworami kanalików łzowych i zwiększoną podatnością na infekcje worków łzowych (zapalenie pęcherza moczowego). (Każdy wewnętrzny róg szpary powiekowej otwiera się na małą przestrzeń zawierającą otwór na kanał łzowy.) pacjenci mogą mieć nienormalnie szeroki, wysoki grzbiet nosa i niedorozwinięte „skrzydełka” nosowe (hipoplazja skrzydełek nosowych), co prowadzi do zwężenia nozdrza. Ponadto w niektórych przypadkach brwi mogą być niezwykle grube i/lub rosnąć razem (sinofreeze). W rzadkich przypadkach osoby dotknięte chorobą mają również szeroko rozstawione oczy (hiperteloryzm oka). Jak wspomniano wcześniej, naukowcy opisali różne formy choroby w oparciu o pewne objawy i określone dane genetyczne. CB typu II różni się od CB typu I brakiem kanału telewizyjnego.
Niektóre osoby z SV mogą mieć dodatkowe nieprawidłowości twarzy. Mogą obejmować:
- niezwykle zaokrąglony czubek nosa, który może być lekko zadarty;
- nienormalna „gładkość” pionowego rowka górnej wargi (rowka);
- pełne usta;
- niewielki występ żuchwy (prognatyzm żuchwy).
Pojawiło się również kilka doniesień, w których choroba była związana z rozszczepem podniebienia (heiloschisis) i / lub nieprawidłowy rowek na górnej wardze (rozszczep wargi).
Zespół Waardenburga często wiąże się z zaburzeniami pigmentacji spowodowanymi niedoborem pigmentu melaniny. U niektórych z tym schorzeniem po urodzeniu występuje siwienie skóry głowy i rzęs (polioza), które z wiekiem zwykle zanika. Brwi, rzęsy i włosy na skórze głowy mogą przedwcześnie stać się szare lub białe (od wczesnego dzieciństwa, okresu dojrzewania lub wczesnej dorosłości). Ponadto niektórzy pacjenci mają obszary skóry o nieregularnym kształcie, pozbawione pigmentacji (leukoderma lub bielactwo nabyte), zwłaszcza na twarzy i dłoniach. Zaburzenie może być również związane z niedorozwojem (hipoplazją) włókien tkanki łącznej, które stanowią większość zabarwionego obszaru obu oczu (tęczówki). W rezultacie pacjenci mogą mieć niezwykle bladoniebieskie oczy lub różnice w pigmentacji tęczówki lub w różnych obszarach tej samej tęczówki (heterochromia tęczówki). Na przykład tęczówka jednego oka może być niebieska, a druga tęczówka innego koloru, albo jedna lub obie tęczówki mogą wydawać się nietypowo „nakrapiane”. Niektóre doniesienia sugerują, że heterochromia tęczówki może występować częściej w zespole Waardenburga typu 2, podczas gdy natomiast obecność siwiejących włosów na skórze głowy i odbarwionych obszarach skóry jest częstsza u osób z zespołem Waardenburga 1 rodzaj.
Przeczytaj także:Hipofosfatazja
Niektóre osoby z FD cierpią również na wrodzoną głuchotę. To upośledzenie słuchu wydaje się być wynikiem nieprawidłowości lub braku narządu Corti, struktury wewnątrz wydrążonego spiralnego kanału ucha wewnętrznego (ślimaka). Narząd Cortiego zawiera maleńkie komórki rzęsate, które przekształcają drgania dźwiękowe w impulsy nerwowe, które są następnie przekazywane przez nerw słuchowy (nerw ślimakowy przedsionkowy) do mózgu. Anomalie w narządzie Cortiego mogą zakłócać przekazywanie tych impulsów nerwowych, powodując upośledzenie słuchu (tzw. niedosłuch odbiorczy lub niedosłuch ślimakowy). U większości pacjentów z tym zaburzeniem wrodzona głuchota czuciowo-nerwowa dotyczy obu uszu (obustronna). Jednak w rzadkich przypadkach może to dotyczyć tylko jednej strony (jednostronnej).
W niektórych przypadkach charakterystyczne cechy twarzy, oczu i słuchu mogą wystąpić w połączeniu z obustronnymi wadami rozwojowymi ramion i dłoni (kończyny górne). Ta forma zaburzenia, opisana jako ciężka manifestacja CB1, jest czasami określana jako zespół Waardenburg typ 3 (CB3), zespół Kleina-Waardenburga lub zespół Waardenburga z anomaliami górnymi odnóża. Wady dwustronne mogą obejmować:
- niedorozwój (hipoplazja) i nieprawidłowe skrócenie kończyn górnych;
- Nieprawidłowe zgięcie niektórych stawów palców w stałych pozycjach (przykurcze zgięciowe)
- połączenie kości nadgarstka;
- i / lub taśma lub fuzja (syndaktylia) niektórych palców.
W niektórych przypadkach mogą występować inne nieprawidłowości szkieletu, takie jak wrodzone wysokie barki (deformacja Sprengla).
Opisano również czwartą postać zespołu Waardenburga, w której główne cechy SV są związane z chorobą Hirschsprunga. Ta forma zaburzenia może być nazwana CB4, zespołem Waardenburga-Schacha lub zespołem Waardenburga-Hirschsprunga. Choroba Hirschsprunga (znany również jako bezzwojowe rozdęcie okrężnicy) jest zaburzeniem żołądkowo-jelitowym charakteryzuje się brakiem niektórych ciał komórek nerwowych (zwojów) w ścianie mięśni gładkich w obszar okrężnicy. W rezultacie dochodzi do braku lub naruszenia mimowolnych rytmicznych skurczów, które napędzają pokarm wzdłuż przewodu pokarmowego (perystaltyka). Powiązane objawy i oznaki mogą obejmować:
- nieprawidłowe nagromadzenie kału w okrężnicy;
- ekspansja okrężnicy nad dotkniętym segmentem (megacolon);
- wzdęcia (bębnica);
- wymioty;
- brak apetytu (anoreksja);
- niezdolność do wzrostu i przybierania na wadze w oczekiwanym tempie;
- i/lub inne odchylenia.
Zgłaszano rzadkie przypadki CB4, w których pacjenci mieli również objawy neurologiczne z powodu nieprawidłowości w mózgu i rdzeniu kręgowym (ośrodkowy układ nerwowy). W takich przypadkach dodatkowe wskazania obejmowały:
- ograniczenie wzrostu;
- nieprawidłowo obniżone napięcie mięśniowe (niedociśnienie);
- przykurcze i deformacje kończyn (arthrogrypoza);
- i/lub inne odchylenia.
Powoduje
Mutacje genów EDN3, EDNRB, MITF, PAX3, SNAI2 oraz SOX10 może powodować zespół Waardenburga. Geny te biorą udział w tworzeniu i rozwoju kilku typów komórek, w tym komórek produkujących pigment zwanych melanocytami. Melanocyty wytwarzają pigment zwany melaniną, który wpływa na kolor skóry, włosów i oczu oraz odgrywa zasadniczą rolę w prawidłowym funkcjonowaniu ucha wewnętrznego. Mutacje w którymkolwiek z tych genów zakłócają prawidłowy rozwój melanocytów, powodując nieprawidłową pigmentację skóry, włosów, oczu i problemy ze słuchem.
Przeczytaj także:Ból gardła
Typy I i III zespołu Waardenburga są spowodowane mutacjami w genie PAX3. Mutacje genów MITF lub SNAI2 może powodować zespół Waardenburga typu II.
Mutacje w geny SOX10, EDN3 lub EDNRB może powodować zespół Waardenburga typu IV. Oprócz rozwoju melanocytów geny te są ważne dla rozwoju komórek nerwowych w okrężnicy. Mutacje w jednym z tych genów prowadzą do utraty słuchu, zmian pigmentacyjnych i problemów jelitowych związanych z chorobą Hirschsprunga.
W niektórych przypadkach genetyczna przyczyna zespołu Waardenburga nie została ustalona.
Rodzaj dziedziczenia
Zespół Waardenburga jest zwykle dziedziczony w sposób autosomalny dominujący, co oznacza, że wystarczy jedna kopia zmienionego genu w każdej komórce, aby wystąpiło zaburzenie. W większości przypadków chory ma jednego rodzica z tą chorobą. Niewielki odsetek przypadków jest wynikiem nowych mutacji w genie; takie przypadki występują u osób, które nie mają rodzinnej historii choroby.
W niektórych przypadkach zespołu Waardenburga typu II i IV stwierdza się autosomalny recesywny wzór dziedziczenia, co oznacza, że obie kopie genu w każdej komórce mają mutacje. Najczęściej rodzice pacjenta z chorobą autosomalną recesywną noszą jedną kopię zmutowanego genu, ale nie wykazują objawów choroby.
Dotknięte populacje
Zespół Waardenburga (SV) nosi imię badacza (Petrus J. Waardenburg), który jako pierwszy dokładnie opisał to zaburzenie w 1951 roku. Od tego czasu w literaturze medycznej opisano co najmniej 1400 przypadków. Dostępne dane wskazują, że choroba może mieć częstość występowania około 1 na 40 000 urodzeń i odpowiada za 2 do 5 procent wrodzonej głuchoty. Choroba dotyka mężczyzn i kobiety stosunkowo w równym stopniu.
Zaburzenia objawowe
Objawy następujących zaburzeń mogą być podobne do objawów zespołu Waardenburga. Porównania mogą być przydatne w diagnostyce różnicowej.
Istnieje wiele zaburzeń, które mogą mieć pewne cechy podobne do tych obserwowanych w SV. Na przykład według naukowców takie zaburzenia mogą obejmować:
- rodzinne przypadki częściowe bielactwo i głuchota;
- rodzinne przypadki bielactwa nabytego i wrodzonej odbiorczej / odbiorczej utraty słuchu;
- stan znany jako zespół Vogta-Koyanagi-Harady.
Te ostatnie mogą charakteryzować się zapalnymi chorobami oczu; bielactwo nabyte; siwienie brwi, rzęs i włosów na głowie (polioza); wypadanie włosów (łysienie); stan, w którym niektóre dźwięki mogą powodować dyskomfort (dyskusja); i/lub inne objawy i oznaki.
Ponadto niektóre wady wrodzone mogą być również związane z bocznym przemieszczeniem wewnętrznego kącika oka (tj. teleskopowym); szeroko rozstawione oczy (hiperteloryzm oka); wąskie nozdrza; niezwykle grube brwi, które mogą rosnąć razem (sinofreeze); upośledzenie słuchu; wady rozwojowe kończyn górnych; zaburzenia trawienia; i / lub inne funkcje potencjalnie związane z zespołem Waardenburga. Jednak takie zaburzenia często charakteryzują się dodatkowymi, charakterystycznymi objawami, objawami fizycznymi lub innymi cechami, które mogą pomóc odróżnić je od SV.
Diagnostyka
Zespół Waardenburga (SV) można zdiagnozować przy urodzeniu lub we wczesnym dzieciństwie (lub w niektórych przypadkach w późniejszym życiu) na podstawie dokładna ocena kliniczna, identyfikacja charakterystycznych objawów fizycznych, pełna historia pacjenta i jego rodziny, a także różnych specjalistycznych Badania. Na przykład u pacjentów z podejrzeniem CO ocena diagnostyczna może obejmować użycie suwmiarki do pomiaru odległości między wewnętrznymi kącikami oczu, zewnętrznymi kącikami oczu a źrenicami (między źrenicami dystans). ( Suwmiarka to instrument z dwoma przegubowymi, ruchomymi, zakrzywionymi ramionami, które służą do pomiaru grubości lub średnicy.) Naukowcy zauważają, że identyfikacja i analiza połączenie tych pomiarów może czasami być przydatne do potwierdzenia obecności lub braku bocznego przemieszczenia wewnętrznego kącika oka, co może wskazywać na obecność SV 1 rodzaj.
Można przeprowadzić dodatkowe testy diagnostyczne, aby pomóc wykryć lub scharakteryzować pewne nieprawidłowości potencjalnie związane z chorobą. Badania takie mogą obejmować badanie pod mikroskopem podświetlanym w celu uwidocznienia wewnętrznych struktur oka (badanie lampą szczelinową); specjalistyczne badania słuchowe; i/lub zaawansowane techniki obrazowania, takie jak ocena anomalii ucha wewnętrznego, wad szkieletu (np. obserwowana w SV typu 3), choroba Hirschsprunga (np. obserwowana w zaburzeniu typu 4) oraz itd.. Na przykład naukowcy podkreślają, że tomografia komputerowa (CT) może pomóc scharakteryzować wady ucha wewnętrznego, które są odpowiedzialne za wrodzoną głuchotę odbiorczą. (Tomografia komputerowa wykorzystuje komputer i promieniowanie rentgenowskie do stworzenia filmu przedstawiającego przekrojowe obrazy struktur wewnętrznych.) W niektórych przypadkach ocena diagnostyczna może również obejmować usunięcie (biopsja) i badanie mikroskopowe niektórych próbek tkanek, takich jak biopsja odbytu, w celu potwierdzenia choroby Hirschspring. W niektórych przypadkach mogą być również zalecane dodatkowe badania diagnostyczne.
Przeczytaj także:Jej choroba
Zabiegi standardowe
Leczenie zespołu Waardenburga ma na celu wyeliminowanie określonych objawów pojawiających się u każdej osoby. Takie leczenie może wymagać skoordynowanego wysiłku zespołu pracowników służby zdrowia, takich jak lekarze skóry (dermatolodzy); okuliści (oftalmolodzy); specjaliści od słuchu; lekarze zajmujący się diagnostyką i leczeniem chorób kośćca, stawów, mięśni i tkanek pokrewnych (ortopedzi); lekarzy specjalizujących się w choroby przewodu pokarmowego (gastroenterolodzy); logopedzi; fizjoterapeuci; i/lub innych pracowników służby zdrowia.
Wczesne rozpoznanie niedosłuchu czuciowo-nerwowego może odgrywać ważną rolę w zapewnieniu szybkiej interwencji i odpowiedniej opieki podtrzymującej. W niektórych przypadkach lekarze mogą zalecić leczenie implantem ślimakowym – urządzeniem, które: w którym elektrody wszczepione do ucha wewnętrznego stymulują nerw słuchowy do wysyłania impulsów do mózg. Ponadto można zalecić wczesną edukację specjalną, aby pomóc w rozwoju mowy i określonych technik. (na przykład język migowy, czytanie z ruchu warg, korzystanie z narzędzi komunikacyjnych itp.), które mogą ułatwić Komunikacja.
Ponieważ osoby z zaburzeniami pigmentacji skóry mogą być predysponowane do oparzeń słonecznych i ryzyka rozwoju raka skóry, lekarze mogą zalecić unikaj bezpośredniego światła słonecznego, używaj kremów z filtrem o wysokim współczynniku ochrony przeciwsłonecznej (SPF), noś okulary przeciwsłoneczne i powłoki ochronne przed słońce. (takich jak czapki, długie rękawy, spodnie itp.) i inne odpowiednie środki. Pacjenci ze zmniejszoną pigmentacją tęczówki, bocznym przemieszczeniem wewnętrznych kącików oczu i/lub inne współistniejące nieprawidłowości oczne, okuliści mogą również zalecić pewne środki wspierające. Mogą one obejmować stosowanie specjalnie przyciemnianych okularów lub soczewek kontaktowych (na przykład w celu zmniejszenia możliwej wrażliwości na światło (światłowstręt)), środki zapobiegania lub leczenia infekcji lub inne środki zapobiegawcze lub terapeutyczne.
U osób z nieprawidłowościami kończyny górnej leczenie może obejmować fizjoterapię i różne techniki ortopedyczne, w tym ewentualnie zabieg chirurgiczny. Ponadto czasami zaleca się operację w celu leczenia innych nieprawidłowości, które mogą być związane z chorobą. Konkretne wykonywane zabiegi chirurgiczne będą zależeć od ciężkości i lokalizacji nieprawidłowości anatomicznych, związanych z nimi objawów i innych czynników.
Na przykład u pacjentów z chorobą Hirschsprunga leczenie może wymagać usunięcia dotkniętego obszaru jelita i chirurgicznego „zjednoczenia” zdrowych obszarów jelita. W niektórych przypadkach, przed chirurgiczną korektą stanu, leczenie może wymagać stworzenia sztuczne ujście okrężnicy przez otwór w ścianie jamy brzusznej (na przykład tymczasowy kolostomia).
Dodatkowe usługi wsparcia, które niektóre ofiary mogą uznać za pomocne, obejmują edukację specjalną i/lub inne usługi medyczne, społeczne lub zawodowe. Poradnictwo genetyczne przyniesie również korzyści osobom dotkniętym chorobą i ich rodzinom. Inne leczenie zespołu Waardenburga jest objawowe i podtrzymujące.
Prognoza
Zespół Waardenburga nie powinien wpływać na długość życia. Zwykle chorobie nie towarzyszą żadne inne powikłania poza utratą słuchu, zaburzeniami pigmentacji czy chorobą Hirschsprunga, która atakuje jelito grube. Cechy fizyczne spowodowane tą chorobą pozostaną z osobą na całe życie.