Zespół Pradera-Williego
 zespół Pradera-Williego - rzadko anomalią genetyczną charakteryzuje zatrzymywania działania genów, ani w inny sposób naukowej, zjawisko to spowodowane brakiem ekspresji, co oznacza przeniesienie informacji genetycznej z DNA na RNA.Funkcją genów w tym przypadku jest włączenie do pracy we właściwym czasie. Na przykład chłopcy nie mają wzrostu włosów na twarzy w dzieciństwie, a zjawisko to zaczyna się po początkowym okresie dojrzewania. Ta choroba została po raz pierwszy opisana w 1956 roku. Zespół
zespół Pradera-Williego - rzadko anomalią genetyczną charakteryzuje zatrzymywania działania genów, ani w inny sposób naukowej, zjawisko to spowodowane brakiem ekspresji, co oznacza przeniesienie informacji genetycznej z DNA na RNA.Funkcją genów w tym przypadku jest włączenie do pracy we właściwym czasie. Na przykład chłopcy nie mają wzrostu włosów na twarzy w dzieciństwie, a zjawisko to zaczyna się po początkowym okresie dojrzewania. Ta choroba została po raz pierwszy opisana w 1956 roku. Zespół
Prader-Willy występuje z częstością jednego przypadku przypadającego na 12 000 niemowląt urodzonych. Ta choroba została opisana w jego pracach Heinrich Willy, Andrea Prader, Andrew Ziegler, Alexis Labhart, Guido Fanzoni.
Zespół Pradera-Williego powoduje
Choroba charakteryzuje się brakiem lub wyrażając siedem genów z chromosomu XV, odziedziczone po ojcu. Zespół Pradera-Williego jest oznaczona przez zmiany w chromosomie ojcowskim, aw przypadku zmiany chromosomie dominującej występuje syndrom Angelmana.
W zestawie genów, który powoduje powstawanie zespołu Pradera-Williego, kopia genu otrzymanego od ojca aktywnie funkcjonuje, a matka nie jest. Oznacza to, że gdy większość ludzi ma jedną roboczą kopię tych genów, pacjenci z zespołem Pradera-Willi żyją bez takiej kopii.
Zespół Pradera-Williego objawy
tej choroby charakteryzuje dysplazji, hipotonia( niskie napięcie mięśniowe), otyłość( tło do przejadania się pojawić w 2. roku).Cierpi z zespołem Pradera-Williego mają obniżoną koordynację ruchów, a także niską gęstość kości, są właściciele małych stóp i rąk, niskiego wzrostu, nachylonych do snu, zeza, mają krzywiznę kręgosłupa. Dla takich osób charakteryzuje się gruba ślina, hipogonadyzm( obniżone funkcje gruczołów płciowych), obecność złych zębów, niepłodność.Pacjenci charakteryzują się upośledzeniem umysłowym, rozwojem mowy, opóźnieniami w dojrzewania i doświadczają trudności z opanowaniem umiejętności motorycznych.i zespół objawów choroby
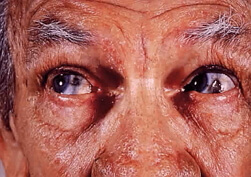
Pradera-Williego wizualnie obserwowano dużą grzbiet nosa, wąskie i wysokie czoło migdałowe oczy, wąskie usta. Wszystkie te objawy nie występują u jednego pacjenta. Często pacjent spotyka się z pięcioma z powyższych objawów.
Diagnoza zespołu Prader-Williego
Ta choroba może być rozpoznana przed urodzeniem. Wskazuje to na małą mobilność płodu, często niewłaściwą pozycję dziecka, jak również wielowodorotwórczość płynu owodniowego w nadmiernych ilościach. Zespół
Prader-Willi jest często diagnozowany po analizie genetycznej. Ta analiza dokonuje noworodka z hipotonią( zmniejszenie napięcia mięśniowego).Czasami lekarze popełniają błędy i diagnozują zespół Downa lub miopatię.Znaki diagnozowania choroby po urodzeniu należą: urodzić poprzez cesarskie cięcie, prezentacji zamka, niedociśnienie tętnicze, senność, słaby odruch ssania z powodu osłabionych mięśni tonu malucha, trudności w oddychaniu, hipogonadyzmu.
Dzieci z zespołem Pradera-Williego są podobne, więc doświadczony genetyk natychmiast diagnozuje chorobę, nawet bez oczekiwania na wyniki badania krwi.
Zespół Pradera-Williego i Angelmana zespół
Dziś wiemy jeszcze jedną chorobę, które odnoszą się do choroby farmacja zespół Pradera-Williego. Ta choroba nazywa się Angelman. Charakteryzuje się mutacją w matczynym materiale genetycznym. Wyniki większości niezależnych badań wskazują na takie przyczyny choroby jako mutacje genu. Produktem tego genu jest enzym składnik całego kompleksowego systemu degradacji białka. Choroba ta pochodzi od pediatry z Wielkiej Brytanii, Harry'ego Angelmanna, który opisał go w 1965 roku.
zespół Pradera-Williego leczoną chorobę
, jako wrodzona genetyczny pozostaje niezbadane do końca i środki traktowania nie działało.
Jak traktować zespół Pradera-Williego? Niemniej jednak istnieją pewne środki medyczne, które poprawiają jakość życia ludzi. Na przykład dzieci z niedociśnieniem wymagają masażu, a także innych typów terapii specjalnej. Pokazuje użycie specjalnych technik poprawiających rozwój dziecka, a także zajęcia z defektologiem i terapeutką mowy. Wskazane są również dzieci w wieku 7, 8, 9 lat z zespołem Pradera-Willi, hormony wzrostu, terapię hormonalną gonadotropinami.
Zespół Pradera-Willy'ego u chłopców przejawia się w hipogonadyzmie, mikro penia, a także w kryptoryzmie( nie owulacji w jądrach).W takiej sytuacji lekarze zalecają lub poczekać, aż testy zejdą lub zalecą operację terapii hormonalnej. Aby poprawić przyrost masy ciała, zaleca się dietę, która ogranicza ilość węglowodanów i tłuszczów. Jeśli otyłość jest już dostępna, należy kontrolować ilość i jakość żywności, która jest wchłaniana przez pacjentów z zespołem Pradera-Williego. Dla takich ludzi jest charakterystyczny wilgotny apetyt. Możliwe powikłanie przebiegu choroby bezdechu, charakteryzuje się opóźnieniem w oddychaniu we śnie.
Prognoza zespołu Prader-Williego
Podczas planowania drugiego dziecka te same rodzice mogą mieć drugie ryzyko tej choroby. To zależy od mechanizmu powodującego nieprawidłowe funkcjonowanie genetyczne. Ryzyko jest mniejsze niż 1% w przypadkach, gdy pierwsze dziecko ma delecję genu lub jednoskładnikowy disenmy partenogenetyczne. Ryzyko wynosi do 50%, pod warunkiem, że nie jest to spowodowane mutacją.Ryzyko do 25% pozostaje przy translokacji chromosomów rodzicielskich. W każdym razie rodzice muszą przeprowadzić badanie genetyczne.
Zespół Pradera-Williego i jego prognoza często zachowują opóźnienie w mowie, a także rozwój psychiczny. Badania przeprowadzone przez Freim i Kerfs wykazały, że 5% pacjentów ma średni współczynnik wywiadu. To odpowiada 85 lub więcej punktom na skali IQ.27% pacjentów ma poziom inteligencji na krawędzi średniej, szacowanej na 70-85 punktów.34% pacjentów ma poziom wywiadu odpowiadający 50-70 punktom.27% pacjentów pozostaje na poziomie średniej luki i szacuje się na 35-70 punktów.5% pacjentów ma silne opóźnienie i osiąga 20-35 punktów, a mniej niż 1% ma znaczne opóźnienie. Inne badania wykazują, że 40% pacjentów wykazuje średnią lub mniejszą inteligencję.
Dzieci z zespołem Pradera-Williego zazwyczaj mają dobrą długoterminową i wizualną pamięć.Są w stanie nauczyć się czytać, mieć pasywne, bogate słownictwo, ale ich własna mowa jest często gorsza niż jej zrozumienie. Pamięć słuchowa, umiejętności pisania i umiejętności matematyczne, krótkotrwała pamięć słuchowa i wizualna również cierpią i nie spełniają standardów wiekowych. Zespół
Prader-Willi często towarzyszy zwiększonemu apetytowi, ponieważ pacjenci mają zwiększone stężenie we krwi poziomu hormonu ghreliny. U pacjentów, typowe jest niskie stężenie somatoliberyny. Niektórzy kojarzy to z 15. chromosomem, który jest związany z podwzgórzem. Inne dane wskazują na zmniejszenie całkowitej liczby komórek, a także komórek zawierających oksytocynę w jądrach parapomórkowych podwzgórza. Co ciekawe, istnieją dowody wskazujące na to, że jest odwrotnie: autopsja zmarłych z tą chorobą często nie wykazuje wad w podwzgórzu.