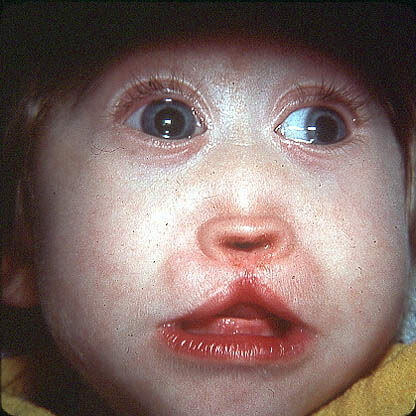
Fibrose cística
 A fibrose cística( do latim «muscus» -. Refere-se ao muco ou viscoso, «viscidus» - pegajosas) - uma doença congénita pesada caracterizada por lesões das glândulas exócrinas e faz com que a patologia do sistema respiratório e digestivo. A mucucovisisose geralmente se desenvolve na infância.
A fibrose cística( do latim «muscus» -. Refere-se ao muco ou viscoso, «viscidus» - pegajosas) - uma doença congénita pesada caracterizada por lesões das glândulas exócrinas e faz com que a patologia do sistema respiratório e digestivo. A mucucovisisose geralmente se desenvolve na infância.
A fibrose cística da criança se manifesta quando mesmo um dos pais é um portador do gene CFTR que sofreu uma mutação. Anualmente em todo o mundo nascem mais de 45.000 crianças com patologia hereditária da fibrose cística. Esta doença é de importância social devido à natureza progressiva do curso e às possíveis mortes subsequentes, bem como à incapacidade de muitos pacientes.
A fibrose cística causa
A mutação genética é a base absoluta desta doença. Se heterocigotos - ambos os pais, então, a possibilidade de produzir uma criança doente com fibrose cística em uma família é equivalente a 25%.E a frequência de transporte nos genes é de 2-5%.
No final do século passado, cientistas genéticos decifraram a estrutura de um gene que é responsável pela combinação de uma proteína. Foi chamado CFTR, o que significa um regulador transmembranar de fibrose cística. Esta proteína regula o movimento de eletrólitos através das membranas das células que alinham os canais de saída das glândulas exócrinas. E por causa da mutação desse gene, há uma ruptura das estruturas e de todas as funções da proteína, de modo que o fluido secretor que as glândulas secretam passa para um conteúdo espesso e viscoso. Como resultado, órgãos internos tão importantes como o pâncreas, o trato gastrointestinal e os pulmões são afetados.
O próprio processo de mutação é caracterizado pela proliferação de um triplete que codifica o aminoácido fenilamina. Uma molécula de um gene mutado com um distúrbio na posição 508 perde o seu resíduo de aminoácido. Assim, apareceu o nome "Delta 508". No momento, já são conhecidas e estudadas mais de 120 espécies de diferentes mutações envolvidas no desenvolvimento de patologias. Um número tão grande deles pode ser explicado em uma variedade de imagens clínicas de fibrose cística.manifestação bastante pesada da doença nos estágios iniciais observados em pacientes F homozigótica 508. pacientes A tendo nenhuma F 508, polimorfismo clínica diferente, isto é, eles simultaneamente diagnosticado com formas graves de doenças, os primeiros sintomas e o resultado adverso com relativamente bons indicadores de doença mais tarde na vida eNa adolescência.
Sintomas de fibrose cística
Existem várias formas principais de fibrose cística - pulmonar, intestinal, respiratória e intestinal. Muito menos comum na prática médica é a obstrução meconial do intestino, edematoso-anêmico e outras formas.
A forma mais grave de manifestação da fibrose cística é a forma mista da doença. Ela atende em 75% dos casos. Na anamnese de tais pacientes, são observados repetidos repetidos tipos graves de bronquite e pneumonia, às vezes mesmo com um curso prolongado.É examinado o quadro clínico de tosse constante, com escarro viscoso segregado, violações freqüentes do trato gastrointestinal. Mas inequivocamente, as mudanças mais graves ocorrem por parte dos órgãos respiratórios. Maior viscosidade selecção das secreções mucosas das glândulas brônquicas mukostaz cria e leva a infecção, o que torna possível a manifestar-se o progresso e a bronquite crónica, em que aparece uma tosse dolorosa característico duro com expectoração viscoso descarregado, além purulenta.
As alterações broncopulmonares são parte integrante da patência prejudicada de todo o sistema brônquico. Alterações que ocorrem como resultado da autolimpação, geralmente causam obstrução de bronquíolos e pequenas partes dos brônquios. O enfisema dos pulmões se desenvolve como resultado do alongamento dos espaços de ar, e devido ao bloqueio completo dos brônquios, todos os sinais de atelectasia são observados. Eles podem ser pequenos e alternar com focos de enfisema.
Em um quadro clínico severo, às vezes com tais mudanças, aparecem micro-insetos que muitas vezes afetam as glândulas submucosas bronquiais. Em seguida, o relâmpago começa a progredir do parênquima pulmonar, que é monitorado por uma forma aguda de pneumonia, que difere em pacientes com curso grave, prolongada da clínica de fibrose cística, propenso a formação de abscessos.
Periodicamente, as primeiras manifestações da forma pulmonar de fibrose cística podem às vezes ocorrer no segundo ou terceiro ano de vida, e também em épocas posteriores. A natureza dessas manifestações adquire todas as formas de pneumonia prolongada e grave.
Os filhos mais velhos são mais propensos a apresentar um quadro clínico de bronquite prolongada com obstrução bem definida. As alterações causadas pelo processo inflamatório são manifestadas por bronquite difusa, que muitas vezes se repete com várias pneumonias de natureza prolongada e depois se transforma rapidamente em inflamações crônicas dos pulmões. Em seguida, desenvolva gradualmente bronquiectasias, bem como pneumosclerose. O tecido intersticial é afetado e esse é o resultado da pneumofibrose generalizada. Portanto, em ambos os pulmões são úmidos, mas, em geral, são escutados relvados borbulhantes pequenos.
Extremamente desfavorável para o curso da fibrose cística é o desenvolvimento de pneumosclerose extensa e bronquiectasias, seguida da formação de um processo purulento. Todos os tipos de alterações obstrutivas observadas na fibrose cística, no curso da progressão, levam a sintomas aumentados de enfisema, uma violação pronunciada da respiração visual e patologias de desenvolvimento da pequena circulação.
Em paralelo com a doença subjacente, a deformação do tórax ocorre, as falanges terminais tornam-se um tipo de varas timpânicas e a insuficiência cardíaca se desenvolve como um coração pulmonar.
Piopnevmotorax, pneumotórax e hemorragia pulmonar raramente são complicações da fibrose cística. E se a doença é caracterizada pela duração do fluxo, observe as lesões nasofaríngeas, que incluem pólipos, adenóides e amigdalite crônica. Quase todas as crianças são diagnosticadas com sinusite, nas manifestações clínicas das quais há uma voz nasal, dores de cabeça e secreção de secreção das passagens nasais.
De acordo com o quadro clínico de distúrbios do intestino, bem como do pâncreas, todos os sintomas de pacientes com fibrose cística das principais formas da doença. Mas as mudanças que podem reduzir a atividade das enzimas do pâncreas, especialmente em crianças transferidas para alimentação artificial, podem se manifestar por fração insuficiente de proteínas e gorduras e também pela absorção. Portanto, no intestino desenvolvem processos de podridão, que levam ao acúmulo de gases e inchaço constante. Como resultado, mesmo com o primeiro exame do paciente, é possível assumir fibrose cística, levando em conta uma fezes abundantes características com odor putrefativo desagradável. Quase 20% dos pacientes têm prolapso retal.
Alguns dos sintomas abdominais são distinguidos por dores frequentes no abdômen de diferentes etiologias. Eles são de tipo cãibras com flatulência, dor nos músculos de uma tosse paroxística prolongada no hipocôndrio direito, na região do fígado.
A localização da dor na região epigástrica pode ocorrer porque o suco gástrico não é suficientemente neutralizado no duodeno. Isso ocorre como resultado da secreção prejudicada do pâncreas. Portanto, em uma grande quantidade de pacientes falecidos que freqüentemente têm resultados bioquímicos negativos, uma autópsia relata cirrose do fígado do tipo biliar. Em pacientes com fibrose cística, observa-se aumento do apetite, mas distúrbios digestivos persistentes levam constantemente a hipotrofia.Às vezes, mas raramente ocorre edema hipoproteinêmico. O desenvolvimento da hipotrofia é simultaneamente promovido pelo desenvolvimento de hipocloremia, anorexia e alcalose metabólica.
Fibrose cística em crianças
Uma vez que a fibrose cística é uma das patologias hereditárias mais comuns para hoje, a freqüência de ocorrência entre crianças de diferentes idades é muito alta. Além disso, sua probabilidade de desenvolvimento não será afetada de forma alguma pela pertença da criança a um determinado sexo. A doença não escolhe - é um menino ou uma menina. A principal coisa a entender é que a fibrose cística em crianças desenvolve-se apenas por defeitos genéticos. Portanto, nenhum mau hábito, status social ou problemas ambientais podem ser uma probabilidade de nascimento de crianças com a patologia da fibrose cística.
As crianças são diagnosticadas com esta doença apenas quando possuem dois genes mutantes que herdaram de seus pais. Portanto, em famílias onde o casal, e ambos, portadores da fibrose cística do gene, nascerão crianças com essa patologia. Mas aqueles que herdaram de seus pais apenas uma espécie desse gene, serão considerados portadores de fibrose cística. Essas crianças nascerão absolutamente saudáveis.
As crianças têm uma grande variedade de manifestações clínicas de fibrose cística. Alguns podem detectar mudanças no sistema respiratório, principalmente doenças brônquicas e pulmonares, outros - doença pancreática grave. O curso clínico da doença de um tipo, para cada criança, pode prosseguir de diferentes maneiras.
É importante lembrar que a fibrose cística é uma patologia genética que não afeta as habilidades mentais da criança.
Nos primeiros meses de vida de muitas crianças, a hipotrofia atua ao mesmo tempo que mantém o apetite e somente depois de algum tempo nas fezes, bem como a poléfala, a gordura é formada, o que preocupa os pais do bebê, então eles prestam atenção especial.
A pele do bebê é uma boa indicação para o diagnóstico, pois tem um sabor "salgado".Portanto, com essa queixa, os pais pela primeira vez consultam um pediatra. Talvez em tais crianças, uma forma acentuada de hipocalemia e alcalose metabólica devido a distúrbios eletrolíticos pronunciados se manifestará.
Na fibrose cística, por parte do sistema respiratório, a tosse torna-se o primeiro sintoma desta doença. No início, ele se manifestará sob a forma de "tosse", depois aumentará gradualmente e assumirá o caráter de tosse como em tosse convulsa. Crianças, cianose e falta de ar, podem acompanhar essa tosse paroxística, mas as paradas respiratórias geralmente não acontecem. Escarro excretado ao tossir, no início da doença, cor clara e não viscosa, mas depois torna-se mucopurulento e adquire uma forte viscosidade. Se houvesse um escarro esverdeado, então este é um sinal sobre a aparência no corpo de uma criança de Pseudomonas aeruginosa e sua infecção pelo escarro. Em muitos casos, o ARVI é freqüentemente registrado, que se repete periodicamente e tem correntes prolongadas, bem como uma tosse que persiste por um longo período de tempo.
Na fase inicial da fibrose cística, a imagem da doença na auscultação de crianças pequenas às vezes é normal. Um exame aprofundado da criança permite detectar um tamanho anteroposterior alargado do tórax, uma leve falta de ar com pouco esforço físico e uma redução da circulação dos pulmões.
Por parte do trato gastrointestinal em crianças do primeiro ano de vida, revela-se uma fezes líquidas freqüentes de cor esverdeada, que posteriormente se torna gordurosa. Uma criança com fibrose cística tem um peso corporal que não corresponde a pares saudáveis, isto é, está atrasado no desenvolvimento físico e, posteriormente, também no crescimento. Crianças com fibrose cística que têm exame visual têm uma massa muscular reduzida, o abdômen abaulando para fora e o prolapso do reto geralmente é um sintoma desta patologia.
Além das formas principais de fibrose cística, por exemplo, 15% dos recém-nascidos desenvolvem obstrução meconial do intestino. Os sintomas deste tipo de patologia podem ser vômitos com a bile, a impossibilidade de divergência do mecônio e um aumento do abdômen. O mecônio cinza é mantido no intestino delgado, na válvula ileocecal. A manifestação mais perigosa desta forma de fibrose cística é a sua complicação na forma de peritonite meconial.
A totalidade de todos esses processos patológicos do lado da respiração, digestão e glândulas sudoríparas, em geral, conduz a distrofia infantil. A hipoproteinemia ea anasarca em lactentes ocorrem em uma combinação de anemia hemolítica. Muitas vezes, essas crianças têm uma deficiência em vitaminas lipossolúveis. Por exemplo, a causa do sangramento é uma deficiência de vitamina K, como conseqüência da hipoprotrombinemia.
Crianças que sofrem de fibrose cística são caracterizadas por uma forte perda de sais na transpiração, como resultado do calor, seios inflamados freqüentes, que são complicados pela polipose e pelo envolvimento da glândula parótida.80% das crianças doentes têm uma sensibilidade aumentada a vários tipos de alérgenos, como alimentos, domicílio e medicamentos.
Uma das complicações da fibrose cística em crianças é a doença celíaca, mas uma complicação freqüente é colelitíase.
Diagnóstico de Fibrose Cística
O diagnóstico de fibrose cística é a base do processo broncopulmonar crônico, síndrome intestinal, teste de andorinha e fibrose cística de irmãos. Portanto, para diagnosticar esta patologia, é suficiente ter duas dessas características listadas. Certos critérios para o diagnóstico de fibrose cística já foram desenvolvidos, que incluem dois tipos de diagnóstico.
O primeiro é baseado nos sintomas clínicos característicos. Isso pode ser tanto uma história familiar de fibrose cística quanto a triagem neonatal de tripsina imunorreactiva de natureza positiva.
O segundo é o aumento do teor de cloretos de potássio( mais de 60 mmol / l), as duas mutações identificadas, um resultado positivo durante a medição da diferença nos potenciais nasais. O diagnóstico é confiável, mesmo que exista um critério de cada tipo de diagnóstico.
Para diagnosticar fibrose cística, recorrer a uma série de métodos bastante informativos, mas também demorados. Para isso, é necessário determinar a concentração de cloreto de sódio no suor, realizar estudos de cólica fecal, fazer diagnósticos de DNA, medir a diferença nos potenciais nasais, determinar a atividade da elastase pancreática em fezes.
Atualmente, existe uma oportunidade real para realizar um exame pré-natal do feto quanto à presença de fibrose cística, em conexão com o diagnóstico de DNA disponível. E é necessário fazer esse exame para futuros pais, mesmo antes do planejamento da gravidez, ou seja, determinar a presença ou ausência de um gene de fibrose cística no corpo.
Até à data, a maioria das famílias que têm uma criança doente com fibrose cística, mas que sofreu esse tipo de diagnóstico como o DNA, tem no futuro crianças já bastante saudáveis.
Análise para fibrose cística
Há uma série de testes, diagnósticos e testes que são utilizados para diagnosticar fibrose cística. Nos primeiros meses de vida em neonatos podem realizar diagnósticos neonatais. Este método consiste em determinar a quantidade de conteúdo IRT no sangue de uma criança.ИРТ é uma enzima do pâncreas. Se uma criança está doente com fibrose cística, a análise em ИРТ será com um índice aumentado. Tal análise é feita se houver suspeitas de fibrose cística.
O teste de diagnóstico mais eficaz para fibrose cística é um teste de suor. O procedimento padrão é que um site de pele é tomado para um teste de suor, no qual a ionoforresis com pilocarpina foi previamente realizada. A concentração de NaCL nas secreções das glândulas sudoríparas não deve exceder 40 mmol / l. Se o resultado da amostra de teste for superior a 60 mmol / l, então pode ser considerado positivo. Um ponto importante é a reanalisação.É feito se a resposta for positiva, duvidosa ou negativa, mas o quadro clínico sugere a probabilidade de ter fibrose cística.
Para finalmente diagnosticar, é necessário realizar vários desses testes, dois ou três e obter um resultado confirmatório positivo.
Mas às vezes as respostas são falsas. Isso pode ser devido à cobrança imprecisa de material e ao seu transporte;Tomando uma amostra de um recém-nascido ou de um paciente tomando cloxacilina.
Entre os métodos de pesquisa, há também métodos de coprologia. Pesquisando pacientes kaproliyu kala, você pode detectar steatorii. Isso se deve ao fato de que o duodeno tem baixa atividade ou ausência completa de enzimas pancreáticas nele.
Quando a radiografia de tórax pode detectar densidades brônquicas, e também observar um aumento na freqüência do tecido pulmonar. A radiografia mostra sinais de queda nos segmentos do pulmão, especialmente o lobo superior. Este é um indicador de fibrose cística.
O uso do estudo das funções de respiração externa permite determinar o quanto o sistema respiratório é afetado. Este exame determina como os brônquios irão reagir aos broncodilatadores e identificar pacientes para quem o propósito desses medicamentos é razoável.
Para crianças mais velhas, o método mais informativo para diagnosticar uma doença é medir a diferença nos potenciais nasais. Sua principal tarefa é encontrar o principal defeito que causa o desenvolvimento da fibrose cística. Os indicadores são retirados da mucosa nasal e da pele do antebraço.
Dois outros tipos de análise podem ser observados - genética e pré-natal. A primeira é uma análise bastante dispendiosa, e a segunda é o diagnóstico de DNA.
Tratamento da fibrose cística
No momento, o medicamento é impotente na cura completa da fibrose cística. Cientistas de muitos países estão trabalhando no desenvolvimento de drogas no nível celular, mas, infelizmente, um resultado positivo até agora não pode ser alcançado. Os médicos só podem esperar que algum dia esta doença seja completamente curada. O método de tratamento mais relevante poderia ser "terapia genética", que seria responsável por entregar cópias saudáveis de um gene mutado para células doentes. Mas estes são apenas pressupostos e tentam tratar a patologia hereditária com métodos conhecidos.
A base para o tratamento da fibrose cística é uma abordagem gradual desta doença. Mais importante ainda, é necessário criar um ambiente favorável, atribuir uma determinada dieta com adição de enzimas, realizar fisioterapia e proporcionar ao paciente uma atividade física adequada.
Para o sucesso da terapia, é necessário observar em um dispensário altamente qualificado e apoio moral de pessoas próximas.
A abordagem terapêutica para o tratamento de um paciente deve ser realizada exclusivamente de forma individual e contínua. Para fazer isso, é importante parar o processo infeccioso que ocorre no sistema respiratório no tempo. Em seguida, restaure a função respiratória e de limpeza dos brônquios. E no final do tratamento, ajuste a deficiência de enzimas no pâncreas.
O processo de tratamento de doenças pulmonares inclui medidas que reduzem a viscosidade do escarro e melhoram a drenagem dos brônquios, bem como a terapia com antibióticos, tratamento de intoxicação, hipoxia, hipovitaminosis e insuficiência cardíaca.
Para reduzir a viscosidade do escarro, prescrevem-se preparações enzimáticas tais como Hymopsin, Chymotrypsin, Fibrinolisina ou fármacos mucolíticos. Comprimidos de Mukosolvin internados e intramuscularmente - acetilcisteína. Você pode usar Bromhexine e Muciltin, mas eles têm um efeito de diluição fraco. Para uma boa drenagem dos brônquios, é necessária a massagem da área torácica dos pulmões e a ginástica terapêutica. Os bebês, usando uma bomba elétrica, produzem escarro. Cada paciente deve ter um inalador de compressor, com o qual é possível introduzir um fármaco eficaz Pulmozim.
Se as doenças pulmonares estão em processo de exacerbação, recorra à consulta do curso de antibioticoterapia por 3-4 semanas. Para fazer isso, eles devem fazer um antibiótico, mas se for impossível determinar, então, com a finalidade dos medicamentos, são tomadas patógenos fibrose cística - Pseudomonas aeruginosa e estafilococos.
Simultaneamente com medicamentos antibacterianos, anti-histamínicos e antimicrobianos são prescritos. No momento da exacerbação da doença, a terapia UHF e a eletroforese de magnésio são feitas. Para reduzir a hipertensão do pulmão, Euphyllin é prescrito em comprimidos a 7-10 mg / kg por dia. Para melhorar o metabolismo do miocardio, os propósitos da Cocarboxilase e do Orotate de Potássio são mostrados. Com um coração pulmonar, eles tomam Digoxina, Glucocorticotides( 1-1,5 mg / kg por dia).
No tratamento da síndrome intestinal, os pacientes recebem novas preparações enzimáticas revestidas com um revestimento e resistentes ao meio ácido - Pancitrat e Creon, que são mais efetivos do que Mezim-forte, Festal ou Panzinorm. Em pacientes com fibrose cística, estes medicamentos devem ser tomados durante toda a vida, e a dosagem é prescrita estritamente individualmente para cada paciente. A dor adequada no abdômen, uma fezes normalizadas e a falta de gordura neutra no exame de fezes para escatologia são consideradas suficientes para que a enzima seja tomada.
Independentemente do processo generalizado e das mudanças que ocorrem em muitos órgãos e sistemas, a observação dispensária de crianças doentes por um médico distrital e um pneumologista é obrigatória. Além disso, os pais devem aprender a cuidar de uma criança doente, realizar suas próprias massagens, ginástica terapêutica e aerossol. O acompanhamento clínico é necessário para controlar as funções dos brônquios, pulmões, coração, trato digestivo, fígado, rins e recepção adequada das preparações enzimáticas, para a provisão oportuna de cuidados médicos para exacerbações da doença, para terapia restauradora geral e para alívio obrigatório de infecções crônicas.
A maioria das crianças com fibrose cística está em atendimento ambulatorial, onde a criança será totalmente provida de cuidados domiciliários e a recaída de infecções pode ser descartada. Somente com formas graves da doença geralmente são pacientes hospitalizados. Isso se refere à insuficiência respiratória do grau II-III, descompensação do coração pulmonar e hemoptise. O íleo meconial, bem como a obstrução intestinal, em que o tratamento conservador não é eficaz, é uma indicação para a intervenção cirúrgica.
Também pode notar-se que existem métodos mais radicais para tratar a fibrose cística, onde consideram o transplante de órgãos de acordo com os sinais vitais. Isto é principalmente praticado em clínicas na Alemanha, Canadá e Estados Unidos. São essas operações que ajudam alguns pacientes com patologia hereditária a sobreviver e viver o tempo suficiente.
Existem, obviamente, métodos de tratamento não convencionais. Mas eles são mais auxiliares e são aplicados no contexto do principal.
Crianças com fibrose cística intestinal leve a moderada podem receber tratamento em um sanatório. Se for possível criar grupos especiais para formas pulmonares da doença, também é útil para essas crianças tomarem tratamento de sanatório. Os critérios para a seleção de pacientes para este tipo de tratamento são desordens intestinais compensadas após a adoção de preparações enzimáticas, violações ausentes no coração pulmonar e todos os tipos de processos inflamatórios.
Não é recomendado para crianças doentes com fibrose cística estarem na creche e na manjedoura. Mas você só pode ir à escola em condições boas ou satisfatórias, mas você ainda deve ter um dia livre fora da semana. Durante o exame ou o tratamento, tais crianças estão completamente isentas de frequentar as aulas e passando todos os exames.
Todas as crianças com diagnóstico de fibrose cística estão em um check-up de saúde para a vida. A única coisa é que aos 15 anos são transferidos para a conta de observação, para o terapeuta.