Lipoidos (Lysosomal lipid storage disease): vad är det, exempel
Uppmärksamhet! Informationen är endast i informationssyfte och är inte avsedd att diagnostisera och förskriva behandling. Rådgör alltid med din specialistläkare!
Innehåll
- Vad är lysosomala lagringssjukdomar?
- Vad är lipider?
- Hur ärfts lipoidos?
- Typer och symtom på lysosomala lagringssjukdomar
Vad är lysosomala lagringssjukdomar?
Lysosomala lagringssjukdomar (lipoidos), är en grupp ärftliga metabola sjukdomar där skadliga mängder fettämnen (lipider) samlas i olika celler och vävnader i kroppen.
Människor med dessa störningar gör antingen inte tillräckligt med ett av de enzymer som behövs för att bryta ner (metabolisera) lipider, eller så producerar de enzymer som inte fungerar korrekt.
Med tiden kan denna överdrivna ansamling av fett leda till permanent skada på celler och vävnader, speciellt i hjärnan, det perifera nervsystemet (nerver från ryggmärgen till resten av kropp), lever, mjälte och benmärg.
Vad är lipider?
Lipider är fettliknande ämnen som är viktiga delar av membranen inuti och mellan celler och i myelinskidan, som täcker och skyddar nerverna. Lipider inkluderar oljor, fettsyror, vaxer, steroider (såsom kolesterol och östrogen) och andra relaterade föreningar.
Dessa fettämnen lagras naturligt i kroppens celler, organ och vävnader. Små kroppar i celler, som kallas lysosomer, omvandlar eller metaboliserar regelbundet lipider och proteiner till mindre komponenter för att ge energi till kroppen. Störningar där intracellulärt material, som inte kan metaboliseras, finns kvar i lysosomer, kallas lysosomala lagringssjukdomar.
Förutom sjukdomar associerade med ackumulering av lipider, inkluderar andra lysosomala sjukdomar associerade med ackumulering mukolipidoser, i vilka celler och vävnader ackumuleras en överdriven mängd lipider med sockermolekyler fästa vid dem, såväl som mukopolysackaridoser, i vilka en överdriven mängd stort, komplext socker ackumuleras molekyler.
Hur ärvs lipoidos?
Lysosomala lagringssjukdomar ärvs från en eller båda föräldrarna som bär på en defekt gen som reglerar ett specifikt lipidmetaboliserande enzym i en klass av celler i kroppen. De kan ärvas på två sätt:
- Autosomalt recessivt arv uppstår när båda föräldrarna bär och vidarebefordrar en kopia av den defekta genen, men ingen av föräldrarna påverkas av sjukdomen. Varje barn som föds av dessa föräldrar har 25 procents chans att ärva båda kopiorna av den defekta genen, 50 procent sannolikheten att han kommer att vara en bärare som liknar sina föräldrar, och en 25 procents chans att inte ärva någon kopia av den defekta gen. Barn av båda könen kan ärvas på ett autosomalt recessivt sätt.
- X-länkad (eller könsrelaterad) recessiv arv uppstår när mamman bär den påverkade genen på X-kromosomen. X- och Y-kromosomerna är involverade i könsbestämning. Kvinnor har två X-kromosomer, medan män har en X-kromosom och en Y-kromosom. Söner till kvinnliga bärare har 50 procents chans att ärva och påverka sjukdomen, eftersom söner får en X-kromosom från sin mamma och en Y-kromosom från sin pappa. Döttrar har 50 procents chans att ärva den påverkade X-kromosomen från sin mamma och är bärare eller lindrigt påverkade. Drabbade män överför inte sjukdomen till sina söner, men deras döttrar kommer att vara bärare och bärare av sjukdomen.
Typer och symtom på lysosomala lagringssjukdomar
Gauchers sjukdom - den vanligaste av lysosomala sjukdomar. Sjukdomen orsakas av en brist på enzymet glukocerebrosidas, vilket leder till ackumulering av glukocerebrosid i många vävnader. Fettmaterial kan ansamlas i hjärnan, mjälten, levern, njurarna, lungorna och benmärgen.
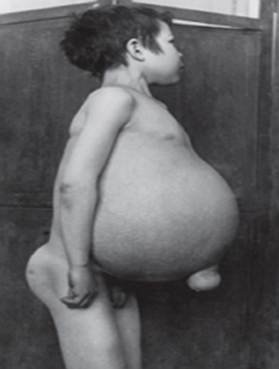
Symtom kan innefatta hjärnskador, förstoring av mjälten och lever, nedsatt leverfunktion, skelettsjukdomar och benskador som kan orsaka smärta och frakturer, svullnad av lymfkörtlar och (ibland) intilliggande leder, uppblåsthet, brunaktig hudton, anemi, lågt antal blodplättar och gula fläckar på ögonen.
De personer som drabbas allvarligast kan också vara mer mottagliga för infektioner. Sjukdomen drabbar män och kvinnor lika.
Gauchers sjukdom har tre generella kliniska undertyper:
- Typ 1 (Eller nej neuronopatisk typ) är den vanligaste formen av sjukdomen i USA och Europa. Hjärnan påverkas inte, men det kan uppstå skador på lungorna och i sällsynta fall njurarna. Symtom kan börja tidigt i livet eller i vuxen ålder och inkluderar en förstorad lever och kraftig förstoring av mjälten, vilket kan leda till bristning och ytterligare komplikationer. Skelettsvaghet och skelettsjukdom kan vara omfattande. Människor i denna grupp får vanligtvis lätt blåmärken på grund av det låga antalet blodplättar i blodet. De kan också uppleva trötthet på grund av anemi. Beroende på sjukdomens uppkomst och svårighetsgrad kan personer med typ 1 kan leva till vuxen ålder. Många lider av mild sjukdom eller kanske inte visar några symtom. Fastän 1typ Gauchers sjukdom är vanlig bland människor av Ashkenazi-judiskt arv och kan drabba människor av alla etniska bakgrunder.
- Typ 2 (eller akut neuropatisk barndom Gauchers sjukdom) börjar vanligtvis inom 3 månader efter födseln. Symtom inkluderar omfattande och progressiva hjärnskador, spasticitet, kramper, stela lemmar, förstorad lever (leverhepatomegali) och mjälte (splenomegali), onormala ögonrörelser och dålig förmåga att suga och svälja. Drabbade barn dör vanligtvis före 2 års ålder.
- Typ 3 (kronisk neuronopatisk form) kan börja när som helst under barndomen eller till och med i vuxen ålder. Den kännetecknas av långsamt progressiva men mindre allvarliga neurologiska symtom jämfört med akut Gauchers sjukdom 2 typer. Viktiga symtom inkluderar ögonrörelserubbningar, kognitiva brister, dålig koordination, kramper, förstoring av mjälte och/eller lever, skelettsjukdomar, blodsjukdomar inklusive anemi och problem med andas. Nästan alla med Gauchers sjukdom 3 typer, som får enzymersättningsterapi kommer att nå vuxen ålder.
För patienter 1:a och 3:e typen Enzymersättningsterapi, som ges intravenöst varannan vecka, kan dramatiskt minska storleken på levern och mjälten, minska skelettavvikelser och vända andra manifestationer. En framgångsrik benmärgstransplantation behandlar de neurologiska manifestationerna av sjukdomen. Denna procedur medför dock betydande risker och görs sällan hos individer med Gauchers sjukdom.
I sällsynta fall kan operation krävas för att ta bort hela eller delar av mjälten (om en person har ett mycket lågt antal trombocyter eller när ett förstorat organ i hög grad påverkar personens komfort). Blodtransfusion kan gynna vissa personer med anemi. Andra kan behöva ledplastik för att förbättra rörligheten och livskvaliteten. Det finns för närvarande ingen effektiv behandling för de hjärnskador som kan uppstå hos personer med Gauchers sjukdom typ 2 och 3.
Niemann-Picks sjukdom är en grupp autosomala recessiva störningar som orsakas av ackumulering av fett och kolesterol i cellerna i levern, mjälten, benmärgen, lungorna och i vissa fall hjärnan.
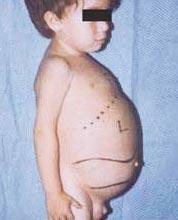
Neurologiska komplikationer kan inkludera ataxi (brist på muskelkoordination som kan påverka promenader, skrivande och ätande, bland andra funktioner), ögonförlamning, hjärndegeneration, inlärningsproblem, spasticitet, svårigheter att äta och svälja, sluddrig tal, förlust av muskeltonus, överkänslighet för beröring och viss opacitet på hornhinnan på grund av överdriven ackumulering material. Hos 50 % av de drabbade utvecklas en karakteristisk körsbärsröd gloria runt mitten av näthinnan, som kan ses av en läkare med ett speciellt instrument.
Niemann-Picks sjukdom klassificeras i tre allmänna undertyper:
- Typ A, den allvarligaste formen, börjar i tidig barndom. Bebisar verkar normala vid födseln, men efter 6 månader utvecklas djupa hjärnskador, en förstorad lever och mjälte och svullna lymfkörtlar och knutar under huden (xantom). Mjälten kan bli 10 gånger sin normala storlek och kan brista och orsaka blödning. Dessa barn blir gradvis svagare, tappar motorisk funktion, kan bli anemiska och är benägna att få återkommande infektioner. De lever sällan mer än 18 månader. Denna form av sjukdomen är vanligast i judiska familjer.
- Typ B(eller juvenil debut) påverkar vanligtvis inte hjärnan, men de flesta barn utvecklas ataxi, skada på nerverna som lämnar ryggmärgen (perifer neuropati), och lungsvårigheter som utvecklas med åldern. Förstoring av lever och mjälte är typiskt för ungdomar. Personer med typ B kan leva relativt länge, men många kräver livslång syrgasbehandling på grund av lungskador. Niemann-Pick typ A och B är resultatet av en ansamling av ett fettämne som kallas sfingomyelin på grund av brist på ett enzym som kallas sfingomyelinas.
- Typ C kan dyka upp tidigt i livet eller utvecklas under tonåren eller till och med vuxen ålder. Niemann-Picks sjukdomtyp C orsakas inte av sflingomyelinasbrist, utan av brist på NPC1- eller NPC2-proteiner. Som ett resultat ackumuleras olika lipider och särskilt kolesterol i nervceller och orsakar deras funktionsfel. Hjärnangrepp kan vara omfattande, vilket resulterar i oförmåga att titta upp och ner, svårigheter att gå och svälja, progressiv hörselnedsättning och progressiv demens. Personer med typ C har endast mild förstoring av mjälte och lever. Förväntad livslängd för personer med typ C varierar avsevärt. Vissa människor dör i barndomen, medan andra som är mindre drabbade också kan leva i vuxen ålder.
Det finns för närvarande inget botemedel mot Niemann-Picks sjukdom. Behandlingen är stödjande. Barn dör vanligtvis av infektion eller progressiv neurologisk förlust. Det har förekommit fall av benmärgstransplantation hos flera patienter med typ B med blandade resultat.
Fabrys sjukdomockså känd som alfa-galaktosidas-A-bristorsakar ackumulering av fettämnen i det autonoma nervsystemet (den del av nervsystemet som kontrollerar ofrivilliga funktioner som andning och hjärtslag), i ögon, njurar och kardiovaskulär systemet.
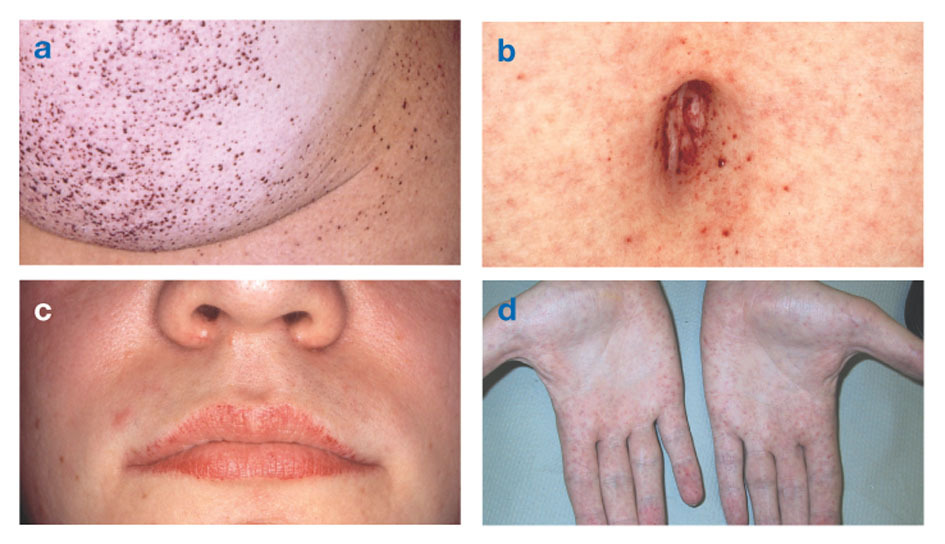
Fabrys sjukdom är den enda X-kopplade lipidsjukdomen. Sjukdomen drabbar främst män, även om en mildare och mer varierande form förekommer hos kvinnor. I sällsynta fall har drabbade kvinnor allvarliga symtom som liknar de som ses hos män med sjukdomen. Symtomdebuten inträffar vanligtvis under barndomen eller tonåren.
Neurologiska tecken inkluderar brännande smärta i armar och ben som blir värre i varmt väder eller efter träning, såväl som ackumulering av överskottsmaterial i hornhinnans transparenta lager (vilket leder till grumling, men utan förändringar i synen). Fettansamling i blodkärlens väggar kan störa cirkulationen och utsätta en person för fara stroke eller hjärtattack. Andra symtom inkluderar ett förstorat hjärta, progressivt njursviktleder till njursjukdom i slutstadiet, gastrointestinala problem, minskad svettning och feber.
Angiokeratom (små, icke-cancerösa, rödlila upphöjda fläckar på huden) kan utvecklas i nedre bålen och bli vanligare med åldern.
Personer med Fabrys sjukdom dör ofta i förtid av komplikationer från hjärtsviktnjursvikt eller stroke. Läkemedel som fenytoin och karbamazepin ordineras ofta för att behandla smärtan som åtföljer Fabrys sjukdom, men inte för att bota den. Metoklopramid eller Lipisorb (kosttillskott) kan lindra gastrointestinala störningar som förekommer ofta hos personer med Fabrys sjukdom, och vissa personer kan behöva en njurtransplantation eller dialys. Enzymersättning kan minska lagringen, lindra smärta och bevara organfunktionen hos vissa personer med Fabrys sjukdom.
Farbers sjukdom också känd som Farbers lipogranulomatosis, beskriver en grupp sällsynta autosomala recessiva störningar som orsakar ackumulering av fettämnen i leder, vävnader och centrala nervsystemet. Det drabbar både män och kvinnor. Sjukdomen börjar vanligtvis i tidig barndom, men kan uppstå senare i livet.
Barn med klassisk Farbers sjukdom utvecklas neurologiska symtom, som kan innefatta ökad sömnighet och slöhet samt problem med svälja. Levern, hjärtat och njurarna kan också påverkas. Andra symtom kan vara ledkontrakturer (kronisk förkortning av muskler eller senor runt lederna), kräkningar, artrit, förstoring lymfkörtlar, ledinflammation, heshet och klumpar under huden som tjocknar runt lederna när den fortskrider sjukdomar.
Drabbade personer med andningssvårigheter kan behöva få en andningsslang insatt. De flesta barn med detta tillstånd dör vid 2 års ålder, vanligtvis från lungsjukdomar. I en av de allvarligaste formerna av sjukdomen kan en förstorad lever och mjälte diagnostiseras kort efter födseln. Spädbarn som föds med denna form av sjukdomen dör vanligtvis inom 6 månader.
Farbers sjukdom orsakas av en brist på ett enzym som kallas ceramidas. Det finns för närvarande ingen specifik behandling för Farbers sjukdom. Kortikosteroider kan ordineras för att lindra smärta. Benmärgstransplantation kan minska granulom (små massor av inflammerad vävnad) hos personer med mild inflammation eller hos patienter utan lung- eller nervsystemskomplikationer. Hos äldre människor kan granulom krympas eller kirurgiskt avlägsnas.
Gangliosidos består av två olika grupper av genetiska sjukdomar. Båda är autosomalt recessiva och påverkar män och kvinnor lika.
GM1 gangliosidos.
GM1-gangliosidos orsakas av en brist på enzymet beta-galaktosidas, vilket resulterar i en onormal ackumulering av sura lipidmaterial, särskilt i nervceller i det centrala och perifera nervsystemet system. GM1 gangliosidos har tre kliniska manifestationer:
- GM1 (den allvarligaste subtypen, som inträffar strax efter födseln) kan inkludera neurodegeneration, kramper, förstorad lever och mjälte, grova ansiktsdrag, oregelbundenheter i skelettet, stelhet i leder, uppblåsthet, muskelsvaghet, överdriven skrämselreaktion och problem med gång. Ungefär hälften av de drabbade får körsbärsröda fläckar i ögat. Barn kan vara döva och blinda vid 1 års ålder och dör ofta vid 3 års ålder av hjärtkomplikationer eller lunginflammation.
- sent barn GM1 gangliosidos börjar vanligtvis mellan 1 och 3 års ålder. Neurologiska symtom inkluderar ataxi, kramper, demens och svårigheter att tala.
- GM1 gangliosidos utvecklas mellan 3 och 30 års ålder. Symtom inkluderar minskad muskelmassa (muskelatrofi), neurologiska komplikationer som är mindre allvarliga och utvecklas långsammare än andra former. störningar, opacitet på hornhinnan hos vissa människor och ihållande muskelsammandragningar som orsakar vridning och repetitiva rörelser eller onormala ställningar (dystoni). Angiokeratom kan utvecklas i den nedre delen av kroppen. Storleken på levern och mjälten är normal hos de flesta drabbade.
GM2 gangliosidos.
GM2 gangliosidos gör också att kroppen ackumulerar överskott av sura fettämnen i vävnader och celler, främst nervceller. Dessa störningar beror på en brist på enzymet beta-hexosaminidas. GM2-störningar inkluderar:
- Tay-Sachs sjukdom (även känd som GM2 gangliosidos variant B) och dess variantformer orsakas av en brist på enzymet hexosaminidas A. Drabbade barn utvecklas normalt under de första månaderna av livet. Symtomen börjar vid 6 månaders ålder och inkluderar progressiv förlust av mental kapacitet, demens, minskad ögonkontakt, ökad en skrämselreaktion på ljud, progressiv hörselnedsättning som leder till dövhet, svårigheter att svälja, blindhet, körsbärsröda prickar på näthinnan och vissa förlamning. Anfall kan börja under det andra året av ett barns liv. Bebisar kan så småningom bli beroende av matningssonden och dör ofta vid 4 års ålder av återkommande infektioner. Tyvärr finns det ingen specialbehandling. Antikonvulsiva medel kan initialt kontrollera anfall. Andra stödjande behandlingar inkluderar rätt näring och hydrering, och metoder för att hålla luftvägarna öppna. En sällsynt form av sjukdomen kallas sent debuterande Tay-Sachs sjukdom, förekommer hos personer mellan 20 och 30 år och kännetecknas av en instabil gång och progressiv neurologisk försämring.
- Sandhoffs sjukdom (Alternativ AB) är en allvarlig form av Tay-Sachs sjukdom. Debut inträffar vanligtvis vid 6 månaders ålder och är inte begränsad till någon etnisk grupp. Neurologiska tecken kan inkludera progressiv försämring av det centrala nervsystemet, motorik svaghet, tidig blindhet, allvarlig rädd reaktion på ljud, spasticitet, chock eller ryckningar muskler (myoklonus), epilepsi, ett onormalt förstorat huvud (makrocefali) och röda fläckar i ögat. Andra symtom kan vara frekventa luftvägsinfektioner, hjärtblåsljud, dockliknande ansiktsdrag och förstoring av lever och mjälte. Det finns ingen specifik behandling för Sandhoffs sjukdom. Liksom med Tay-Sachs sjukdom inkluderar stödjande vård att hålla luftvägarna öppna hela tiden, såväl som korrekt näring och hydrering. Antikonvulsiva medel kan initialt kontrollera epilepsi (kramper).
Krabbes sjukdom(också känd som globulär cell leukodystrofi och galaktosylceramidlipidos) är en autosomal recessiv sjukdom som orsakas av en brist på enzymet galaktocerebrosidas. Sjukdomen drabbar oftast spädbarn som debuterar före 6 månaders ålder, men kan uppstå under tonåren eller vuxen ålder.
Ansamlingen av osmält fett påverkar tillväxten av nervens skyddande hölje (myelinhölje) och orsakar allvarlig försämring av mentala och motoriska färdigheter. Andra symtom inkluderar muskelsvaghet, nedsatt förmåga hos musklerna att sträcka ut (hypotoni), muskelspänningar (spasticitet), plötslig chock eller ryckningar i armar och ben (myokloniska anfall), irritabilitet, oförklarlig feber, dövhet, blindhet, förlamning och svårigheter att svälja. Långvarig viktminskning kan också förekomma.
Sjukdomen kan diagnostiseras genom enzymtestning och identifiering av dess karakteristiska cellgruppering till globoida kroppar i hjärnans vita substans, demyelinisering av nerver och degeneration och förstörelse av hjärnceller. Hos spädbarn är sjukdomen vanligtvis dödlig före 2 års ålder. Personer med en mer avancerad form av sjukdomen har ett lindrigare sjukdomsförlopp och lever betydligt längre.
Ingen specifik behandling för Krabbes sjukdom har utvecklats, även om tidig benmärgstransplantation kan hjälpa vissa människor. Personer med en mer avancerad form av sjukdomen har ett lindrigare sjukdomsförlopp och lever betydligt längre.
Metakromatisk leukodystrofi, eller MLD, är en grupp av störningar som kännetecknas av ackumulering av fettämnen i den vita substansen i centrala nervsystemet och i perifera nerver och till viss del i njurarna. I likhet med Krabbes sjukdom påverkar MLD myelinet, som täcker och skyddar nerverna. Denna autosomala recessiva störning orsakas av en brist på enzymet arylsulfatas A. Denna sjukdom drabbar både män och kvinnor.
Metakromatisk leukodystrofi har tre karaktäristiska former: sent spädbarn, juvenil och vuxen.
- Late Infant MLD börjar vanligtvis mellan 12 och 20 månader efter födseln. Barn kan verka normala till en början, men har svårt att gå och har en benägenhet att falla, åtföljd av intermittent smärta i armar och ben. progressiv synförlust som leder till blindhet, utvecklingsförseningar och förlust av tidigare förvärvade milstolpar, nedsatt sväljning, kramper och demens upp till 2 år. Barn utvecklar också progressiv muskelförtvining och svaghet och förlorar så småningom sin förmåga att gå. De flesta barn med denna form av sjukdomen dör vid 5 års ålder.
- Juvenil MLD börjar vanligtvis mellan 3 och 10 år. Symtom inkluderar försämrad skolprestation, psykisk funktionsnedsättning, ataxi, kramper och demens. Symtomen utvecklas, med döden inträffar 10–20 år efter sjukdomsdebut.
- Vuxen form. Symtom vuxna debut efter 16 års ålder och kan inkludera ataxi, kramper, onormala skakningar i armar och ben (skakningar), försämrad koncentration, depression, psykiska störningar och demens. Döden inträffar vanligtvis 6–14 år efter symtomdebut.
MLD kan inte botas. Behandlingen är symtomatisk och stödjande. Benmärgstransplantation kan i vissa fall försena utvecklingen av sjukdomen. Betydande framsteg har gjorts med genterapi i djurmodeller med MLD och i kliniska prövningar.
Wolmans sjukdomockså känd som lysosomalt surt lipasbristär en allvarlig lipidlagringsstörning som vanligtvis är dödlig vid 1 års ålder. Denna autosomala recessiva störning kännetecknas av en ansamling av kolesterolestrar (vanligtvis transportformen av kolesterol) och triglycerider (den kemiska form i vilken fetter finns i kroppen), som kan ackumuleras avsevärt och orsaka cellskador och tyger.
Både män och kvinnor lider av denna sjukdom. Spädbarn är normala och aktiva vid födseln, men utvecklar snabbt progressiv mental funktionsnedsättning, en förstorad lever och kraftigt förstorad mjälte, uppblåsthet, gastrointestinala problem. gulsot, anemi, kräkningar och kalciumavlagringar i binjurarnafår dem att stelna.
En annan typ lysosomalt surt lipasbrist är kolesterolesterlagringssjukdom. Denna extremt sällsynta sjukdom uppstår som ett resultat av lagring av kolesterolestrar och triglycerider i blodkroppar, lymf och lymfoid vävnad. Hos barn i vuxen ålder förstoras levern, vilket leder till cirros och kronisk leversvikt. Barn kan också ha kalciumavlagringar i binjurarna, och gulsot kan utvecklas i de senare stadierna av sjukdomen.
För närvarande studeras frågan om enzymersättning aktivt både vid Wolmans sjukdom och vid ackumulering av en kolesterolester.
Uppmärksamhet! Informationen är endast i informationssyfte och är inte avsedd att diagnostisera och förskriva behandling. Rådgör alltid med din specialistläkare!