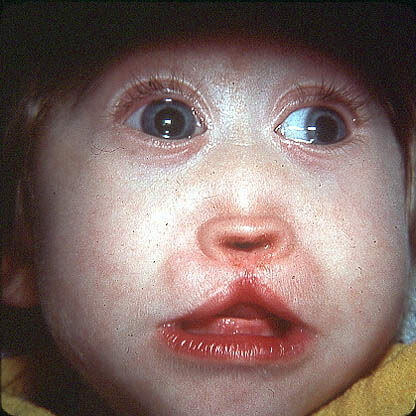
Cystická fibróza
 Cystická fibróza( z latinského «muscus» -. Vztahuje se na sliznici nebo slizký, «viscidus» - lepivé) - těžkým vrozeným onemocněním vyznačující se tím, lézemi exokrinních žláz a způsobuje patologii dýchací a trávicí soustavy. Mucoviscidóza se často rozvíjí v dětství.
Cystická fibróza( z latinského «muscus» -. Vztahuje se na sliznici nebo slizký, «viscidus» - lepivé) - těžkým vrozeným onemocněním vyznačující se tím, lézemi exokrinních žláz a způsobuje patologii dýchací a trávicí soustavy. Mucoviscidóza se často rozvíjí v dětství.
Dítě má cystickou fibrózu, je evidentní, když ani jeden z rodičů je nositelem genu CFTR mutací vystaveny. Každý rok na celém světě více než 45.000 narozených dětí s dědičnou patologii cystickou fibrózou. Tato nemoc má společenskou hodnotu v souvislosti s progresivním průběhem přírody a případných následných úmrtí a invalidity u mnoha pacientů.Cystická fibróza způsobuje
mutace genů - je absolutní základem onemocnění.Pokud heterozygotní - oba rodiče, pak je možnost nemocné dítě s cystickou fibrózou narození v takové rodině se rovná 25%.A frekvence přepravy v genech je 2 až 5%.
na konci minulého století, vědci Genetics dešifrována strukturu genu odpovědného za sloučeninu proteinu. To bylo nazýváno CFTR, což znamená transmembránový regulátor cystické fibrózy. Tento protein reguluje pohyb elektrolytů přes buněčné membrány, které lemuje vylučovací kanálech exokrinních žláz. A v důsledku mutace tohoto genu je porušením všech struktury a funkce proteinů, tak sekreční kapalina, která se izoluje žlázy zhoustne, viskózní obsah. Výsledkem je, že to má vliv takové důležité orgány jako slinivky, zažívacího traktu a plic.
Proces mutace je charakterizován ztrátou tripletu, který kóduje fenylamin aminokyselin. Molekula mutovaného genu, která má poruchu v poloze 508, ztrácí aminokyselinový zbytek. Tímto způsobem, a tam byl takový název - delta 508. V současné době jsou již známy a studoval více než 120 různých typů mutací, které se podílejí na rozvoji chorob. Takový obrovský počet je možno vysvětlit v řadě klinických obrazů cystické fibrózy. Docela těžké projevem onemocnění v raných fázích pozorované u pacientů homozygotní F 508. nemocných, které nemají F 508, odlišný klinický polymorfismus, to znamená, že současně s diagnózou těžkými formami onemocnění, časné příznaky a nežádoucím účinkem s relativně dobrými indikátory onemocnění v pozdějším životě aV dospívání.fibróza příznaky
cystickou
Existuje několik základních forem cystické fibrózy - plicní a střevní, dýchací a střevní.Mnohem méně pravděpodobné, že se nacházejí v lékařské praxi ileus meconium, edematous-anemický a jiných formách.
nejzávažnější formou projevu cystické fibrózy je považován za smíšený forma nemoci. Setká se v 75% případů.Historie těchto pacientů poznamenal opakovaně opakované závažné typy zánětu průdušek a plic, a někdy dokonce s dlouhou historií.Viděl klinický obraz přetrvávající kašel s viskózní sputum přidělené, častému porušování gastrointestinálního traktu. Ale nejednoznačně nejzávažnější změny na straně dýchacích orgánů.Větší viskozita výběr slizničních sekretů z bronchiálních žláz mukostaz Vytváří a vede k infekci, a proto je možné projevovat a pokrok, chronické bronchitidy, v nichž se zdá, charakteristickou bolestivé kašel s tvrdým vypouštěné viskózní sputa, hnisavých kromě.Změny
Bronchopulmonální jsou nedílnou součástí celého bronchiální poruchy průchodnosti systému. Změny, které se vyskytují v důsledku samočištění, mají sklon způsobovat ucpávání průdušinek a průdušek malých dílů.Rozvoji rozedma plic natažením vzduchové mezery a díky dokončení ucpávání průdušek všechny sledované příznaky, atelektázy. Mohou být malé a střídavě s ohnisky emfyzému.
S těžkým klinickým obrazem někdy současně s těmito změnami se objeví microabscesses, které často ovlivňují bronchiální submukózní žlázy. Potom se blesk začne postupovat z plicního parenchymu, která je sledována akutní forma zápalu plic, který se liší u pacientů s těžkou, vleklou průběhu cystické fibrózy klinice, náchylný k abscesu. Pravidelně
prvním projevem plicních cystické formy může někdy nastat na prvním nebo druhém roce života, a k pozdějšímu datu. Povaha těchto projevů získává všechny formy přetrvávající a těžké pneumonie.
Starší děti s větší pravděpodobností předkládají klinický obraz prodloužené bronchitidy s dobře definovanou obstrukcí.Změny způsobené zánětlivým procesem se projevují difúzní bronchitidou, která se velmi často objevuje s různými pneumoniemi s dlouhou dobou přírody a rychle se přeměňuje na chronické záněty plic. Pak se postupně rozvíjí bronchiektáze, stejně jako pneumoskleróza. Intersticiální tkáň je ovlivněna a je výsledkem rozšířené pneumofibrózy. Proto jsou v obou plicích vlhké, ale častěji se naslouchají malé bublinky.
Výrazně nepříznivý průběh cystické fibrózy je vývoj rozsáhlé pneumokleózy a bronchiektázie, po níž následuje vznik hnisavého procesu. Všechny druhy obstrukčních změn pozorovaných v průběhu cystické fibrózy v průběhu progrese vedou ke zvýšení symptomů emfyzému, výraznému narušení vizuálního dýchání a patologickým vývojům malého oběhu.
Paralelně se základním onemocněním dochází k deformaci hrudníku, terminální falangy se stávají jakousi tympanickou tyčinkou a srdeční selhání se vyvíjí jako plicní srdce.
Piopnevmotorax, pneumotorax a plicní krvácení jsou zřídka komplikace cystické fibrózy. A jestliže je onemocnění charakterizováno trváním toku, pak si všimněte nasofaryngeálních lézí, které zahrnují polypy, adenoidy a chronickou tonzilitidu. Téměř všechny děti jsou diagnostikovány sinusitidou, v klinických projevech se vyskytuje nosní hlas, bolesti hlavy a sekrece sekrece z nosních cest.
Podle klinického obrazu poruch střeva a pankreatu jsou všechny symptomy pacientů s cystickou fibrózou hlavních forem onemocnění.Změny, které mohou snížit aktivitu enzymů pankreatu, zejména u dětí přenášených na umělé krmení, se mohou projevit nedostatečným štěpením bílkovin a tuků a také jejich absorpcí.Proto se ve střevě vyvinou hnilobné procesy, které vedou k akumulaci plynů a konstantnímu nadýmání.Výsledkem je, že i při prvním vyšetření pacienta lze předpokládat cystickou fibrózu, přičemž se vezme v úvahu charakteristická hojná stolice s hnilobným nepříjemným zápachem. Téměř 20% pacientů má rektální prolaps.
Některé abdominální příznaky se vyznačují častými bolestmi v břiše různých etiologií.Jedná se o křeče s plynatostí, bolest ve svalech z prodlouženého paroxysmálního kašle v pravém hypochondriu, v oblasti jater.
Lokalizace bolesti v epigastrické oblasti může nastat, protože žaludeční šťáva není dostatečně neutralizována v dvanáctníku. K tomu dochází v důsledku poškození sekrece pankreatu. Proto u velkého počtu zesnulých pacientů, kteří často mají negativní biochemické výsledky, v pitvě hlásí cirhóza jater biliárního typu. U pacientů s cystickou fibrózou je zaznamenána zvýšená chuť k jídlu, ale trvalé trávicí poruchy neustále vedou k hypotrofii. Někdy, ale velmi vzácně se objevuje hypoproteinemický edém. Vývoj hypotrofie je současně podporován rozvojem hypochloremie, anorexie a metabolické alkalózy.
Cystická fibróza u dětí
Vzhledem k tomu, že cystická fibróza je dnes jedním z nejčastějších dědičných onemocnění, frekvence výskytu u dětí různého věku je velmi vysoká.Navíc jeho pravděpodobnost vývoje nebude žádným způsobem ovlivněna příslušností dítěte k určitému pohlaví.Nemoc se nevybírá - je to chlapec nebo dívka. Hlavní věc je pochopit, že cystická fibróza u dětí se vyvíjí pouze genetickými vadami. Proto žádné špatné návyky, sociální status nebo ekologické problémy nemohou být pravděpodobností vzniku dětí s patologií cystické fibrózy.
Děti jsou diagnostikovány touto chorobou pouze tehdy, když mají dva mutantní geny, které zdědily od svých rodičů.Proto se v rodinách, kde jsou dva a dva nosiče genové cystické fibrózy, narodí děti s touto patologií.Ale ti, kteří zdědili od svých rodičů pouze jeden druh takového genu, budou považováni za nosiče cystické fibrózy. Takové děti se budou narodit naprosto zdravé.
Děti mají širokou škálu klinických projevů cystické fibrózy. Některé mohou detekovat změny v respiračním systému, zejména bronchiální a plicní nemoci, jiné - závažné onemocnění slinivky břišní.Klinický průběh onemocnění jednoho typu pro každé dítě může probíhat různými způsoby.
Je důležité si uvědomit, že cystická fibróza je genetická patologie, která neovlivňuje duševní schopnosti dítěte.
V prvních měsících života pro mnoho dětí, špatná výživa stojí při zachování chuti a teprve po nějaké době, tuk ve stolici, stejně jako polifekalie který hlídá grudnichka rodiče, takže dát mu zvláštní pozornost.
Kůže dítěte je dobrým ukazatelem diagnózy, protože má "slanou" chuť.Proto s takovou stížností se rodiče poprvé poradí s pediatrem. Možná, že u takových dětí se projeví ostrá forma hypokalémie a metabolické alkalózy v důsledku výrazných poruch elektrolytů.
Při cystické fibróze se na dýchací cestě stává kašlem prvním příznakem této nemoci. Na začátku, bude to projeví v podobě „kašle“, pak postupně zvyšovat a vzít na charakteru kašle jako černý kašel. Děti, cyanóza a dýchavičnost mohou doprovázet tento paroxysmický kašel, ale přestávky na dýchání obvykle nedochází.Vydáno na kašel hlenu, na počátku onemocnění, světlé a viskózní, ale v budoucnu se stane mukopurulentní a viskozita se stává silnější.Pokud by došlo k nazelenalé sputum, pak je to signál o vzhledu dítěte v těle Pseudomonas aeruginosa infekce a jeho vykašlávání.V mnoha případech se často zaznamenává ARVI, které se pravidelně opakují a mají dlouhé proudy, stejně jako kašel, který trvá dlouhou dobu.
V počáteční fázi cystické fibrózy je obraz onemocnění při auskultaci malých dětí někdy normální.Pečlivé vyšetření dítěte umožňuje detekovat větší velikosti anteroposteriorní hrudi, dušnost s malou nevýznamnou fyzické zátěže a snížení plicní exkurze.
část gastrointestinálního traktu u kojenců odhalit řídká stolice nazelenalou barvu, která se později stane mastný vzhled. Dítě s cystickou fibrózou mají tělesnou hmotnost neodpovídá nepostiženými vrstevníky, která zaostává ve fyzickém vývoji a pozdější růst. Děti s cystickou fibrózou, vizuální kontrolou se sníží svaly, žaludek, vystupující směrem ven, a rektální prolaps často příznakem tohoto onemocnění.
Kromě základních forem cystické fibrózy, například, 15% pacientů vyvinout neonatální mekonia ileus. Symptomy tohoto typu patologie mohou být zvracení žluč, nemožnost divergence mekonia a zvětšení břicha.Šedé mekonium se uchovává v tenkém střevě, v ileocekálním ventilu. Nejnebezpečnějším projevem této formy cystické fibrózy je její komplikace ve formě mekoniální peritonitidy.
Kombinace všech těchto patologických procesů na straně dýchacího ústrojí, trávicího ústrojí a potních žláz obvykle vede k dětské podvýživy. Hypoproteinemie a anasarca u kojenců se vyskytují v kombinaci s hemolytickou anémií.Často mají tyto děti nedostatek vitamínů rozpustných v tucích. Například příčinou krvácení je nedostatek vitaminu K v důsledku hypoprothrombinémie.
děti trpící cystickou fibrózou, a vyznačují se prudkým ztrátou soli při pocení, v důsledku tepla, časté zánět vedlejších dutin nosních, které jsou složité a polypóza léze příušní žlázy.80% nemocných dětí má zvýšenou citlivost na různé typy alergenů, jako jsou potraviny, domácí a léčivé přípravky.
Jednou z komplikací cystické fibrózy u dětí je celiakie, ale častou komplikací je cholelitiáza.
Cystická fibróza diagnóza
diagnózu cystické fibrózy je základem chronické bronchopulmonární procesu, střevní syndrom, pozitivní test potu a cystickou fibrózou sourozenců.Proto diagnostikovat tuto patologii postačí mít dvě z uvedených charakteristik. Byly již vyvinuty určité kritéria pro diagnostiku cystické fibrózy, které zahrnují dva diagnostické typy.
První je založena na charakteristických klinických příznacích. To může být jak rodinná anamnéza cystické fibrózy, tak i neonatální screening imunoreaktivního trypsinu pozitivní povahy.
druhý - vysoký obsah chloridů v potu( 60 mmol / l), které lze identifikovat dvě mutace, zatímco pozitivní nosní potenciální měření rozdílu. Diagnostika je spolehlivá, i když existuje jedno kritérium pro každý typ diagnózy.
Pro diagnostiku cystické fibrózy se využívá řada metod, které jsou poměrně informativní, ale také časově náročné.K tomu je nutné stanovit koncentraci chloridu sodného v potu, provádět studie fekální kolik, provádět diagnostiku DNA, měřit rozdíl v nasálních potenciálech, stanovit aktivitu pankreatické elastázy ve stolici.
V současné době existuje reálná příležitost provést přednatální vyšetření plodu na přítomnost cystické fibrózy v souvislosti s dostupnou diagnostikou DNA.A je třeba provést toto vyšetření i pro budoucí rodiče ještě před plánovaným těhotenstvím, tj. Pro určení přítomnosti nebo nepřítomnosti genu cystické fibrózy v těle.
K dnešnímu dni mají většina rodin, které mají nemocné dítě s cystickou fibrózou, ale prošly takovou diagnostikou jako DNA, v budoucnu děti již docela zdravé.Analýza
pro cystickou fibrózu
Existuje celá řada testů, diagnostik a testů, které se používají k diagnostice cystické fibrózy. V prvních měsících života mohou novorozenci provádět neonatální diagnostiku. Tato metoda spočívá v určení obsahu IRT v krvi dítěte. IRT je enzym pankreatu. Pokud je dítě nemocné cystickou fibrózou, analýza na IRP bude mít zvýšený index. Taková analýza se provádí, pokud existují podezření na cystickou fibrózu.
Nejúčinnějším diagnostickým testem pro cystickou fibrózu je test potu. Standardní postup spočívá v tom, že místo kůže se odebere pro test potu, na kterém byla předtím provedena ionoforéza s pilokarpinem. Koncentrace NaCl v sekrech potních žláz by neměla překročit 40 mmol / l. Je-li výsledek zkušebního vzorku vyšší než 60 mmol / l, pak může být považován za pozitivní.Důležitým bodem je re-analýza. Je-li odpověď kladná, pochybná nebo negativní, klinický obraz naznačuje pravděpodobnost vzniku cystické fibrózy.
Pro konečnou diagnostiku je nutné provést několik takových testů, dva nebo tři a získat potvrzující pozitivní výsledek.
Ale někdy jsou odpovědi nepravdivé.Může to být způsobeno nepřesným sběrem materiálu a jeho přepravou;Vzorek od novorozence nebo pacienta užívajícího cloxacilin.
Mezi metodami výzkumu jsou také metody koprogramů.Při vyšetřování pacientů s kaproliyu kala můžete detekovat steatorii. To je způsobeno skutečností, že duodenum má nízkou aktivitu nebo úplnou nepřítomnost pankreatických enzymů v něm.
Při snímání hrudníku může být zjištěna bronchiální hustota a také pozorováno zvýšení vzdušnosti plicní tkáně.Rádiografie vykazuje známky poklesu segmentů plic, zejména horního laloku. To je ukazatel cystické fibrózy.
Využití studie funkcí vnějších dýchacích funkcí umožňuje určit, do jaké míry je dýchací soustava ovlivněna. Toto vyšetření určuje, jak průdušky reagují na bronchodilatátory a identifikují pacienty, u nichž je účel těchto léků rozumný.
Pro starší děti je nejdůležitějším způsobem diagnostiky onemocnění měření rozdílu v nasálních potenciálech. Jeho hlavním úkolem je najít hlavní vadu, která způsobuje vývoj cystické fibrózy. Indikátory jsou odebírány z nosní sliznice a z kůže předloktí.
Dále je možné zaznamenat dva typy analýz - genetické a prenatální.První je poměrně nákladná analýza a druhá je diagnostika DNA.
Léčba cystické fibrózy
V současné době je lék bezmocný při úplné léčbě cystické fibrózy. Vědci z mnoha zemí pracují na vývoji léků na buněčné úrovni, ale bohužel zatím nelze dosáhnout pozitivního výsledku. Lékaři mohou jen doufat, že jednoho dne bude tato nemoc úplně vyléčena. Nejdůležitějším způsobem léčby by mohla být "genová terapie", která by byla zodpovědná za poskytování zdravých kopií mutovaného genu nemocným buňkám. Ale to jsou jen předpoklady a snaží se léčit dědičnou patologii se známými metodami.
Základem léčby cystické fibrózy je postupný přístup k této nemoci. A co je nejdůležitější, je nutné vytvořit příznivé prostředí, přiřadit určitou stravu přidáním enzymů, provádět fyzioterapii a poskytnout pacientovi odpovídající fyzickou aktivitu.
pro úspěšný výsledek léčby bude potřebovat sledování vysoce dispenzární a morální podporou blízkými.
terapeutický přístup k léčbě pacienta musí probíhat výhradně individuálně a plynule. Na tomto důležitém okamžiku zastavit infekční proces, který probíhá v dýchacím traktu. Poté obnovte funkci dýchacích cest a vymývání průdušek. A na konci léčby upravte nedostatek enzymů v pankreatu. Proces
léčba plicních chorob zahrnuje ty události, které snižují viskozitu sputa a zlepšení bronchiální odvodňování a antimikrobiální terapii, léčbu intoxikace, hypoxie, nedostatky vitamínů a srdečního selhání.
Pro snížení viskozity sputa podávány enzymových přípravků, jako jsou himopsina, chymotrypsin, přípravků nebo Fibrinolizin mukoleticheskogo akce. Tabletové Mukosolvin perorálně a intramuskulárně - Atsetiltsitein. Můžete také použít Bromhexin a Mukaltin, ale mají malý řídnutí účinek. Pro dobrou drenážní průdušek potřeba masážního hrudní a plicní fyzioterapie. Děti, používající elektrické čerpadlo, vytvářejí sputum. Každý pacient by měl mít inhalátor kompresoru, pomocí kterého můžete zadat účinný lék Pulmozyme.
Pokud plicní onemocnění jsou v procesu zhoršení, pak se uchýlit k použití antibiotické terapie v průběhu 3-4 týdnů.K tomu, aby se ujistil antibiotikogrammy, ale pokud to není možné určit, pak se jako základ pro podávání těchto léků jsou převzaty patogeny cystická fibróza - Pseudomonas aeruginosa a Staphylococcus aureus.
Současně s antibakteriální léky předepsané antihistaminika a anti-mikrobiální.V době exacerbace onemocnění je UHF-terapie a elektroforéza s hořčíkem. Pro snížení hypertenze plicní Eufillin podávány v tabletách 7-10 mg / kg za den. Pro zlepšení myokardiální metabolismu znázorněno cílovou kokarboksilazu a orotát draselný.Pro plicní srdce se digoxin, Glyukokortikotdy( 1-1,5 mg / kg za den).
léčba pacientů střevní syndrom předepsaných nové enzymové přípravky potažené a odolný vůči kyselým prostředím - to pantsitrat a Creon, který účinně Mezim forte, sváteční nebo panzinorm.pacientů s cystickou fibrózou, tyto léky je třeba brát celý život, a dávka se jmenuje přísně individuálně pro každého pacienta. Stačí, aby se enzym je považován za vyhynulého bolesti břicha, normalizuje stolice a jeho nedostatek neutrálního tuku ve studiu výkalů na scatology.
Bez ohledu na to zobecněné procesy a změny, které se vyskytují v mnoha orgánech a systémech je povinná lékařská prohlídka nemocných dětí a místního lékaře pulmologem. Kromě toho rodiče potřebují naučit, jak pečovat o nemocné dítě, aby provedla vlastní masáže, fyzioterapii a aerosol. Klinický dohled je třeba ovládat funkce na práci průdušek, plic, srdce, trávicího traktu, játrech, ledvinách a správný příjem enzymatických přípravků pro včasnou lékařskou péči v akutních exacerbací onemocnění, pro posílení terapii, jakož i pro povinné úlevu od chronických infekcí.
Obecně platí, že děti s cystickou fibrózou jsou na ambulantní léčbu, kde bude dítě plně opatřené domácí péči, a lze vyhnout opakující se infekce. Pouze u závažných forem onemocnění jsou obvykle hospitalizovaní pacienti. To platí pro dechové nedostatečnosti II-III stupně, plicní srdeční dekompenzace a hemoptysis. Mekonium ileus a střevní neprůchodnost, při které žádný účinný konzervativní léčby, je indikací pro chirurgický zákrok.
mohou také vzít na vědomí, že existuje radikální léčby cystické fibrózy, který je považován za transplantaci orgánu pro ukazatele zdraví.To se prakticky provádí na klinikách v Německu, Kanadě a Spojených státech. Jsou to právě tyto operace pomáhají přežít někteří pacienti s dědičnými chorobami a žít dostatečně dlouho ještě.
Existují samozřejmě nekonvenční metody léčby. Ale jsou více pomocné a používají se na pozadí hlavního.
Děti s mírnou až střední střevní cystickou fibrózou mohou dostat léčbu v sanatoriu. Pokud je možné vytvořit speciální skupiny pro plicní onemocnění onemocnění, je také užitečné, aby takové děti mohly léčit sanatorium. Kritéria pro výběr pacientů pro tento typ léčby jsou kompenzované střevní poruchy po užívání enzymových přípravků, chybějící poruchy v plicním srdci a všechny druhy zánětlivých procesů.
Nedoporučuje se u nemocných dětí s cystickou fibrózou být v mateřských školách a v jeslích. Ale můžete jít do školy pouze v dobrém nebo uspokojivém stavu, ale musíte mít ještě jeden den mimo týden. Během vyšetření nebo léčby jsou tyto děti zcela osvobozeny od povinné školní docházky a absolvování všech vyšetření.
Všechny děti s diagnostikovanou cystickou fibrózou jsou po celou dobu zdravotní prohlídky. Jediné je, že ve věku 15 let jsou převedeni na pozorovací účet, terapeutovi.