Ufuldkommen osteogenese
 Ufuldkommen osteogenese er en medfødt sygdom i knogler såvel som individuelle bindevævsstrukturer. Et andet navn på ufuldkommen osteogenese er en sygdom i skrøbelige knogler. Sygdommen overføres ved arv, dvs. Er genetisk bestemt.
Ufuldkommen osteogenese er en medfødt sygdom i knogler såvel som individuelle bindevævsstrukturer. Et andet navn på ufuldkommen osteogenese er en sygdom i skrøbelige knogler. Sygdommen overføres ved arv, dvs. Er genetisk bestemt.
Ufuldkommen osteogenese forekommer hos både mænd og kvinder, forekomsten er 1 nyfødt pr. 12 000-15 000 børn. Denne patologi blev først nævnt i det 17. århundrede. Ved ufuldkommen osteogenese forstyrres vækst, hudlæsioner, muskelvæv, blodkar, tænder, sener og høreorganer.
På trods af vanskelighederne for personer med ufuldkommen osteogenese fører de fleste patienter en fuldendt livsstil. I øjeblikket bliver behandlingen af ufuldkommen osteogenese mere og mere populær. Ikke desto mindre er det stadig umuligt at helbrede en patient fuldstændigt med en sådan sygdom.
Osteogenesis imperfecta Osteogenesis imperfecta forårsager
henviser til en heterogen arvelig sygdom, der påvirker bindevæv. Sygdommen er beskrevet af osteopeni og nogle andre kliniske tegn på osteogenese.
Hovedårsagen til denne patologiske udvikling er en arvelig mutation af kollagengener. Sjælden er den spontane forekomst af mutationer i generne af dette protein. Konsekvensen af genmutation er en overtrædelse af kollagensyntese, således patologierne i dannelsen af knogle- og bruskvæv.
Utilstrækkelig mængde syntetiseret kollagen( ikke muteret) er også en af grundene til udviklingen af ufuldkommen osteogenese. I dette tilfælde fortsætter sygdommen i en mere mild form. Ufuldkommen osteogenese af denne art udtrykkes i individuelle frakturer i lemmerne, efter puberteten reduceres antallet af brud som regel.
Moderne medicin skelner mellem forskellige typer ufuldkommen osteogenese under hensyntagen til radiografiske, kliniske, kollagenproteogene ændringer.
Ufuldkomne osteogenesetyper:
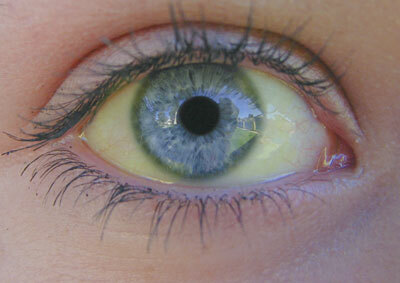
Type I - En svagt udtrykt form, en dominerende type arv. Ufuldkommen osteogenese, der er karakteriseret ved skøre knogler, tilstedeværelsen af blå sclera.
Type II - perinatal-dødelig.
Type III - skeletets deformation er progressiv.
Type IV - den dominerende type arv, deformationerne af skeletet udtages ikke kraftigt, sclera er normalt.
Mange mener, at ufuldkommen osteogenese udvikler sig på baggrund af kvalitative og kvantitative krænkelser af kollagen type I syntese. Ufuldkommen osteogenese af type I er karakteriseret ved et reduceret niveau af syntese af normalt kollagen. Type II, Type IV som osteogenesis imperfecta grund af lavere den totale mængde af kollagen på grund af reduceret stabilitet, mens collagensyntese fortsætter processen uden forstyrrelser.
Ufuldkommen osteogenese symptomer
Hver type ufuldkommen osteogenese har sin karakteristiske symptomatologi.
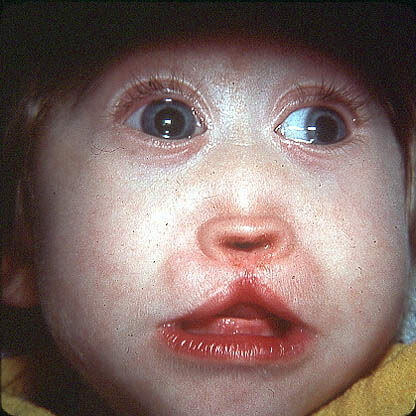
Så jeg skriver ufuldkommen osteogenese hos børn manifesteret af osteoporose, såvel som hyppige brud på knogler. Efter en 10-årig alder falder forekomsten af brud, men stiger igen efter 40 år. Den blå sclera og den tidlige udseende af senilfælgen er noteret. Patienter( ca. 50% af tilfældene) med denne type ufuldkommen osteoporose med lav vækst, nogle af dentin forbliver normale, nogle patienter har dentin opal. I type I forekommer i visse tilfælde ændre aorta, der er mitral( 20% af tilfældene forekommer mitralklapprolaps), næseblod.
Type II af ufuldkommen osteogenese er karakteriseret ved fosterdød før fødslen eller tidlig nyfødt død. Frakturer er flere og hyppige, forekommer let. Tildele 3 grupper:
gruppe A. Skader på hovedet, er lemmer af fosteret under graviditeten noteret på grund af skrøbelighed bindevævsdannelser. Hjernens område af kraniet er forstørret, thoraxen er tværtimod reduceret, lemmerne er korte og buede. Der er alvorlige tilfælde af forkalkning af aortavæggene og endokardiet. Sådanne børn fødes meget lille vækst( ca. 25-30 cm).
Fødsler før sigtet, med 20% af sådanne tidlige fødsler - dødsfald. Nogle syge børn dør i de tidlige dage, nogle lever op til den fjerde uge.kan påvises Patologiske ændringer allerede inden barnets fødsel ved hjælp af røntgenundersøgelse: lår bred, har bølget kanter, kanterne med en rosenkrans, bryst kort. Ved udførelse af medicinsk genetisk rådgivning i proband er der mulighed for en molekylær defekt kombineret med heterozygothed af mutationer lokaliseret i collagengenet. Arven er autosomal dominerende.
gruppe B. fænotypisk ligner gruppe A. Men de krænkelser i luftvejene er ikke så udtalt, børn med osteogenesis imperfecta leve i flere år. Der er forkortede rørformede knogler og ændringer i strukturen af ribbenene, men deres brud er et sjældent fænomen. Formentlig er arvelighed autosomal recessiv.
Gruppe B. Sådanne tilfælde af ufuldkommen osteogenese er sjældne. Karakteriseret ved dødsfald eller dødsårsag i den første måned. Hos patienter er lille vækst, finhed af de rørformede knogler( især diafyse) knogler af kraniet uden nedbrydning noteret. Formentlig en autosomal recessiv type arv.
Patienter med type III imperfekt osteogenese er sjældne. Under fødslen sker brudstykker undertiden, forkortelsen af den nyfødte forkortes, mens massen kan være inden for normale grænser. O-formet deformation af frie ekstremiteter er kyphoscoliosis karakteristisk. Dødsårsagen til omkring halvdelen af patienterne er forskellige ændringer i skelet og kredsløbssystemet. Osteoporose er udtalt, væksten af knogler i længden er brudt sammen med deres synkronisering. I knoglevævets vækstzoner observeres ujævn forkalkning, hvilket fører til forekomsten af spotting( de såkaldte "kornkerner").Arvelighed er ikke ligefrem kendt.
IV type ufuldkommen osteogenese hos børn manifesteres oftest som en lidelse i skeletet. Denne type har en stor variabilitet af osteopeni, antallet af knoglefrakturer, alder, blueness sclera. Sædvanligvis med alderen, er det antal knoglebrud reduceret, dannede hård hud, efter 30 år hos nogle patienter denne type osteogenesis imperfecta hørehæmmede. Ifølge dentin tilstand er patienterne opdelt i 2 grupper: nogle har skarpe ændringer i dentin( opal tænder), andre har ingen ændringer i dentin.
behandling af osteogenesis imperfecta osteogenesis imperfecta
Diagnosen er primært baseret på det kliniske billede af sygdommen. For at etablere en nøjagtig diagnose er en specialisthøring( endokrinolog, ortopædlæge eller genetiker) og udnævnelse af laboratorietests til udelukkelse af andre sygdomme nødvendige.
Laboratoriemetoder omfatter molekylær analyse af kollagen og dets biokemiske analyse, undersøgelse af materialet i hudbiopsi. Instrumentelle metoder af undersøgelsen omfatter: X-ray inspektion( definition af osteopenia, forskellige frakturer, krumning af knogler, brud på rygsøjlen, intercalary knogler i kraniet), biopsi af osteodensiometri.
vigtigste mål for behandling af osteogenesis imperfecta - reducere antallet af frakturer hos patienter, øget knoglemineralisering for at reducere deres skørhed, stigning i knoglemasse. Et vigtigt behandlingspunkt er tilpasningen af barnet såvel som af hans forældre til den ændrede livsstil på grund af sygdom. Dette vil undgå fremtidige brud på knogler.
Hele behandlingsforløbet består af:
- Medicin. For at bekæmpe osteogenesis imperfecta tilførte medikamenter: bisphosphonater, væksthormon, vitamin D3, calciumgluconat, magnesiumsalte og kaliumsalte, ergocalciferol, chelatorer, glycerophosphat.
- Operativ indgriben. Det er nødvendigt at fjerne frakturer, styrke knogler, korrigere forandringer på grund af deformitet, proteser. Sommetider er kirurgisk behandling ikke en operation: På grund af væksten af knogler, der tidligere er indsat, kræver udskiftning.
Ved behandling af frakturer anvendes fleksible titanstænger, som har hypoallergeniske egenskaber. Sådanne stænger er fikseret fraktureret godt, men behovet for yderligere immobilisering af kropdelen er elimineret. Den sædvanlige brug af gipsforbindelser medfører problemer i pleje, det forårsager nye brud. Det er også muligt at anvende forskellige transplantater, knogleplaster og andre metalstrukturer.
lyset af den stigende skrøbelighed af knogler i en patient, i nogle tilfælde, at ortopædisk producere osteoclasis korrigere deformation af knogleændringer, så benet er fastgjort med gips bandage. Også egnet til immobilisering af strækningen af kroppen.
over de grundlæggende fremgangsmåder til behandling af osteogenesis imperfecta anvendes også: udøve terapi, fysioterapi, osteosyntese. Gennemført et kursus af rehabilitering og psykologhjælp, som patient og familiemedlemmer de.
fuldt behandlingsforløb ikke fuldt ud klare sådan alvorlig sygdom som osteogenesis imperfecta, så alle de ovennævnte metoder tillader kun delvist fjerne symptomerne og lette livsstil til patienten uden at påvirke den underliggende årsag til sygdommen.