progeria
 Progeria er en sjælden genetisk sygdom, først beskrevet af Guildford, som manifesteres ved for tidlig ældning af kroppen, der er forbundet med dens underudvikling. Progeri er klassificeret i et barn kaldet syndromet Hutchinson( Hutchinson) -Gilford og adult-Werner syndromet.
Progeria er en sjælden genetisk sygdom, først beskrevet af Guildford, som manifesteres ved for tidlig ældning af kroppen, der er forbundet med dens underudvikling. Progeri er klassificeret i et barn kaldet syndromet Hutchinson( Hutchinson) -Gilford og adult-Werner syndromet.
Med denne sygdom er der en markant retardation i vækst fra barndommen, en ændring i hudens struktur, kakeksi, fraværet af sekundære seksuelle egenskaber og hår, underudvikling af indre organer og udseendet af den gamle mand. I dette tilfælde svarer patientens mentale tilstand til alderen, den epifysiske bruskede plade lukker tidligt, og kroppen har barnets proportioner.
Progeri henviser til uhelbredelige sygdomme og er årsagen til udseendet af alvorlig atherosclerose, som som følge heraf udvikler slagtilfælde og forskellige hjertesygdomme. Og som følge heraf fører denne genetiske patologi til et dødeligt udfald, dvs. Det er fatalt. Som regel kan et barn leve i gennemsnit tretten år, selv om der er tilfælde med en forventet levetid på mere end tyve år.
Pediatrisk Hutchinson-Guildford
Denne sygdom er yderst sjælden i forholdet 1: 4 millioner af nyfødte i Holland og 1: 8 millioner i USA.Desuden påvirker sygdommen flere drenge end piger( 1,2: 1).
Overvej to former for Hutchinson-Guilford progeri: klassisk og ikke-klassisk.
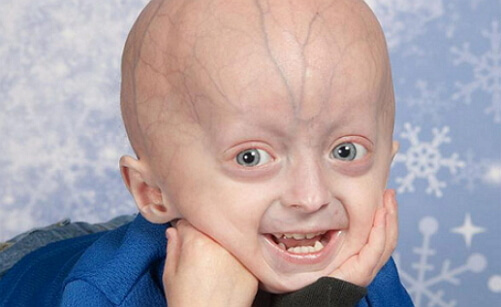
I øjeblikket beskrives mere end hundrede tilfælde af børneprogeri. Og dybest set påvirker denne sygdom børnene i den hvide race. For Hutchinson-Guilford-progerien er en polymorf læsion karakteristisk. Børn, der har dette syndrom, ser helt normale ud ved fødslen. Men allerede et år eller to er der en alvorlig tilbagegang i væksten. Sædvanligvis varierer sådanne børn for lille vækst og endog lavere kropsvægt afhængigt af dens længde.
For børn med progeri er kendetegnet ved fuldstændig skaldethed, ikke kun i hovedbunden, men også i mangel på øjenvipper, øjenbryn fra en tidlig alder. Huden ser svag og rynket ud som følge af det absolutte tab af subkutant fedt, der er cyanose i huden. Hovedet er kendetegnet ved de uforholdsmæssige kraniofaciale knogler, der ligner ansigtet af en fugl med en kroget næse, en unormalt lille underkæbe, bukede øjenbuer og fremspringende ører. Disse funktioner, et stort skaldet hoved og en lille kæbe, giver udseendet af barnet udseendet af en gammel mand.
Andre kliniske manifestationer af progeri inkluderer: unormal og senere tænder, tynd og høj stemme, pæreformet thorax og nedsat størrelse kraveben. Lemmerne er normalt tynde, og de modificerede ulnar- og knæleddene giver det syge barn en "rytterposition".
Børn selv før året har skleraltætninger, medfødt eller erhvervet natur, på balder, hofter og i underlivet. Børn med progeri er præget af hyperpigmentering af huden, som kun stiger med årene og hypoplasien af neglene, hvor de bliver gule, tynde og konvekse, der minder om urglaset. Men siden en alder af fem udvikles en udbredt form for aterosklerose med en stor læsion af aorta og arterier, især mesenterisk og koronar. Og meget senere vises hjertemormer og hypertrofi i hjertet i venstre ventrikel. Tidlig forekomst af aterosklerose hos børn bliver årsagen til deres korte liv. Men hovedårsagen til døden er myokardieinfarkt.
Med progeri er tilfælde af iskæmisk slagtilfælde kendt. Sådanne børn i mental udvikling er absolut ikke forskellige fra sunde børn, nogle gange endda foran dem. Børn med denne diagnose lever i gennemsnit omkring fjorten år.
Med et barnprogeri af en ikke-klassisk form lider kroppens længde lidt over lang tid, håret vedvarer, og lipodystrofi skrider langt langsommere;En recessiv type arv er mulig.

børneprogeri foto
Progeria forårsager
Indtil nu er de nøjagtige årsager til forekomsten af progeri ikke blevet afklaret. Presumptiv ætiologi af udviklingen af denne sygdom er en overtrædelse af metabolisme i bindevæv som følge af proliferation af fibroblaster ved celledeling og øget kollagenproduktion med reduceret syntese af glycosaminoglycaner. Langsom dannelse af fibroblaster skyldes forstyrrelser i det intercellulære stof.
Årsagerne til barndommen progeria syndrom anses mutationer i LMNA gen, som er ansvarlig for kodning lamin A. Dette er et protein, der udgør et af lagene i cellemembranen af kernen.
I mange tilfælde progeria forekommer sporadisk, og i nogle familier, der findes i søskende, især når blodbeslægtede ægteskaber, og dette tyder på en mulig autosomal recessiv form af arv. Når patienter med hudstudier blev detekteret celler, hvori nedsat evne til at fastsætte frakturer og skader på DNA og spiller en genetisk homogene fibroblaster ændre atrofiske epidermis og dermis, der bidrager til forsvinden spæk. For voksne
progeria kendetegnet autosomal recessiv arv af et defekt gen med ATP-afhængig helicase eller WRN.Der er spekulationer i kæden forbinder krænkelser mellem DNA-reparation og udveksling af bindevæv.
fandt også, at Hutchinson-Gilford progeria har uregelmæssigheder i celler bærere, som ikke fuldt ud kan slippe af DNA tværbindinger forårsaget af kemiske midler. Når diagnosticere disse celler findes med dette syndrom, at de ikke er i stand til at gå helt gennem processen med division.
I 1971 Olovnikov blevet foreslået til kortere længder af telomerer under celledannelse. Og i 1992, det er allerede blevet bevist hos patienter med progeria syndrom for voksne. Analyse, der binder Hayflick grænse, telomerlængde og telomeraseaktivitet af enzymet, gør det muligt at kombinere den naturlige ældningsproces til dannelse kliniske symptomer barn Hutchinson-Gilford progeria. Da denne form for Progeria er yderst sjældent, kan vi kun hypotesen om den type arv, som har ligheder med Cockayne syndrom er manifesteret ved de enkelte funktioner i for tidlig aldring.
Der er også udsagn om tilbehør Hutchinson-Gilford progeria til mutation, autosomal dominant, som er opstået de novo, dvs.uden arv. Hun blev en indirekte bekræftelse af syndromet, som er baseret på måling af telomerer kom i bærere af sygdommen, deres forældre og donorer.
Progeria
symptomer Den kliniske billede af børns Progeria forskellige typiske præmatur aterosklerose, myocardial fibrose, sygdomme i cerebral cirkulation, en stigning i lipoprotein og cholesterolniveauer, protrombintid assays, tidlige angreb hjerte, skeletabnormiteter. I dette tilfælde er der givet udtryk for ubalancer ansigt og kranie, kæbe og tænder hypoplasi, hofte forskydning. Lange knogler i normal cortexstruktur og progression af perifer demineralisering udsat tilbagevendende patologiske frakturer.
leddene karakteriseret ved stram mobilitet, især knæet med mulige kontrakturer i hofte, ankel, albue og håndled. Røntgenundersøgelse afslørede demineralisering omkring leddene med osteoporose, varus og valgus deformitet af de nedre lemmer. Også meget ofte udvikler hævelse og fortykkelse af kollagenfibre.
Werner syndrom eller voksen progeria vises mellem 14 og 18 år og er kendetegnet ved en forsinkelse i vækst, grånende universel parallel progression af alopecia.
Typisk progeria syndrom udvikler sig efter tyve år og er kendetegnet ved en tidlig hårtab, udtynding af huden i ansigtet og ekstremiteter, karakteristisk bleghed. Under huden er for stramme synlige overfladiske blodkar og subkutant fedt og muskler placeret nedenunder, helt atrophied, så selvfølgelig ser det uforholdsmæssigt tynd. Så
huden over knogle fremspring bliver gradvist tykkere og ulcerate. Efter tredive år hos patienter med progeria udvikle grå stær i begge øjne, stemmen bliver svag, høj og hæs, væsentligt påvirker huden. Dette kommer til udtryk i form af ændringer sklerotsermopodobnyh ekstremiteter og ansigt, tør hud, sår på fødderne, ligtorne på fødderne og telangiectasia. Sådanne patienter er generelt af lav højde, med lunoobraznym ansigt næbformede næse som en fugl, en indsnævret mundingsåbning og den udragende hage skarpt fuld tynd krop og lemmer.
progeria patienter overtrådt funktion af sved og talgkirtler. Knogle dannes på fremspringene hyperkeratose manifesteret hyperpigmentering almindelighed ændrer formen af neglepladerne. Og efter forskellige skader på ben og fødder vises trofiske sår. Ud over atrofi og udtynding, patienter viser betydelige ændringer i muskler og knogler, forkalkning, generaliseret osteoporose, slidgigt med erosioner. Sådanne patienter er begrænsede i fingers bevægelser og fleksionskontrakt. For patienter med progeria kendetegnet ved deformation af knoglerne som ved reumatoid arthritis, smerter i ekstremiteterne, flade fødder og osteomyelitis. Under
på rentenografii undersøgelse viste knogle osteoporose, heterotopisk forkalkning af huden og subkutane væv, ledbånd og sener. Også, langsomt udvikler grå stær, åreforkalkning udvikler, forstyrret aktiviteterne i det kardiovaskulære system. I de fleste patienter reduceres intellektet.
Efter fyrre år til Progeria diabetes mellitus, dysfunktion af biskjoldbruskkirtlerne og andre sygdomme i næsten 10% af patienterne udvikler tumorpatologi som osteogent sarkom, astrocytoma, thyroid adenocarcinom, brystcancer og hud.
Lethal udfald er normalt en konsekvens af kardiovaskulære patologier og maligne tumorer.
Histologisk analyse af progeria syndrom sæt atrofi af hududvækster, hvor ekkrinnye kirtel tilbageholdte;hvor dermis har fortykkelse gialiniziruyutsya af kollagenfibre, og nervefibre ødelægges.
Patienterne fuldstændig atrofi musklerne, der er ikke noget subkutant fedt.
Sygdommen diagnosticeres på baggrund af kliniske symptomer på progeri. Ved tvivl om diagnosen, evnen af fibroblaster at proliferere i kultur( at falde Werner syndrom).At tage højde for den differentielle diagnose af progeria syndrom Hutchinson-Gilford, Rotmunda-Thomson og systemisk sklerose.
Progeria behandling
Indtil videre er der ingen specifik behandling for progeria, har det endnu ikke udviklet. Grundlæggende terapi er symptomatisk med komplikationer efter forebyggelse af aterosklerose, og i helbredelsen af venøse sår, diabetes.
En anabole effekt er ordineret til STH, som hos nogle patienter øger kropsvægt og længde. Hele behandlingsprocessen udføres en række specialister, såsom en endokrinolog, internist, kardiolog, onkolog, og andre, afhængigt af de fremherskende symptomer.
Men i 2006 noterede amerikanske forskere progression i behandlingen af progeri som en uhelbredelig sygdom. De har gjort til kulturen af fibroblaster beskadigede farnesyltransferaseinhibitor, der tidligere var blevet testet på kræftpatienter. Og denne proces returnerede aldringscellerne til normal form. Dette stof er blevet godt overført, så nu er der håb om, at det vil være muligt i sin anvendelse i fremtiden, for at forhindre snydere selv i barndommen.
Lonafarniba Efficiency( farnesyltransferaseinhibitor) er at øge mængden af fedt under huden, legemsvægt, knoglemineralisering, hvilket i sidste ende reducerer frakturer.
Men alligevel, mens denne sygdom er karakteriseret ved ugunstige prognoser. I gennemsnit lever patienter med progeri i en alder af 13, som dør af blødninger og hjerteanfald.