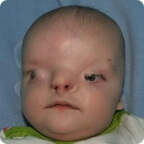
Zystische Fibrose
 Cystische Fibrose( aus dem lateinischen «muscus.» - Bezieht sich auf Schleim oder schleimige, «viscidus» - sticky) - einer schweren angeborenen durch Läsionen der exokrinen Drüsen gekennzeichnet Krankheit und verursacht Pathologie der Atemwege und des Verdauungssystems. Mukoviszidose entwickelt sich häufig in der Kindheit.
Cystische Fibrose( aus dem lateinischen «muscus.» - Bezieht sich auf Schleim oder schleimige, «viscidus» - sticky) - einer schweren angeborenen durch Läsionen der exokrinen Drüsen gekennzeichnet Krankheit und verursacht Pathologie der Atemwege und des Verdauungssystems. Mukoviszidose entwickelt sich häufig in der Kindheit.
Die zystische Fibrose eines Kindes manifestiert sich, wenn selbst einer der Eltern ein Träger des CFTR-Gens ist, das eine Mutation durchgemacht hat. Jährlich werden in der ganzen Welt mehr als 45.000 Kinder mit erblicher Pathologie der zystischen Fibrose geboren. Diese Krankheit ist von gesellschaftlicher Bedeutung aufgrund der fortschreitenden Natur des Kurses und der möglichen nachfolgenden Todesfälle sowie der Behinderung vieler Patienten.
Zystische Fibrose verursacht
Gen-Mutation ist die absolute Grundlage dieser Krankheit. Wenn heterozygot - beide Elternteile, dann ist die Möglichkeit, ein krankes Kind mit zystischer Fibrose in einer solchen Familie zu produzieren, gleich 25%.Und die Häufigkeit des Wagens in den Genen beträgt 2-5%.
Am Ende des letzten Jahrhunderts entschlüsselten Genetiker die Struktur eines Gens, das für die Kombination eines Proteins verantwortlich ist. Es hieß CFTR, was einen zystischen Fibrose Transmembranregler bedeutet. Dieses Protein reguliert die Bewegung von Elektrolyten durch die Membranen von Zellen, die die Ausströmkanäle der exokrinen Drüsen bilden. Und aufgrund einer Mutation dieses Gens ist eine Verletzung der alle Strukturen und Funktionen des Proteins, so sekretorische Flüssigkeit, die Drüsen dick wird, viskosen Inhalt isoliert ist. Infolgedessen sind solche wichtigen inneren Organe wie die Bauchspeicheldrüse, der Magen-Darm-Trakt und die Lunge betroffen.
Der Mutationsvorgang selbst ist durch die Proliferation eines Tripletts charakterisiert, das für die Aminosäure Phenylamin kodiert. Ein Molekül eines mutierten Gens mit einer Störung an Position 508 verliert seinen Aminosäurerest. So erschien der Name "Delta 508". Im Moment sind bereits mehr als 120 Arten verschiedener Mutationen bekannt, die an der Entwicklung von Pathologien beteiligt sind. Eine so große Anzahl von ihnen kann in einer Vielzahl von klinischen Bildern der zystischen Fibrose erklärt werden. Ganz schwere Manifestation der Krankheit in den frühen Stadien bei Patienten homozygot F gesehen 508. A-Patienten, die keine F 508 verschiedenen klinischen Polymorphismus, das heißt, sie gleichzeitig mit schweren Formen von Krankheiten diagnostiziert, frühe Symptome und negativen Ergebnis mit relativ guter Indikator für die Krankheit im späteren Leben undIn der Adoleszenz.
Cystische Fibrose Symptome
Es gibt einige grundlegende Formen der Mukoviszidose - Lunge und Darm, Atemwege und Darm. Viel weniger häufig in der medizinischen Praxis ist Meconium Obstruktion des Darms, ödematös-anämischen und anderen Formen.
Die schwerste Form der Manifestation der zystischen Fibrose ist die gemischte Form der Krankheit. Sie trifft sich in 75% der Fälle. Bei der Anamnese solcher Patienten werden wiederholt wiederholt schwere Arten von Bronchitis und Pneumonie festgestellt, manchmal sogar mit einem längeren Verlauf. Das klinische Bild eines konstanten Hustens, mit sezerniertem viskosem Sputum, häufige Verletzungen des Magen-Darm-Traktes untersucht. Aber unmissverständlich treten die schwersten Veränderungen seitens der Atmungsorgane auf. Größere Auswahl Viskosität von Schleimabsonderungen aus den Bronchialdrüsen mukostaz es schafft und führt zu einer Infektion, die es möglich, zu manifestieren und zu einer chronischen Bronchitis macht, in dem sie eine charakteristische schmerzhaften Husten mit harten entladen viskos Auswurf erscheint, eitrige neben.
Bronchopulmonale Veränderungen sind ein integraler Bestandteil der beeinträchtigten Durchgängigkeit des gesamten Bronchialsystems.Änderungen, die durch Selbstreinigung auftreten, verursachen in der Regel ein Verstopfen von Bronchiolen und kleinen Teilen der Bronchien. Emphysem der Lunge entsteht durch die Streckung von Lufträumen, und durch völlige Verstopfung der Bronchien werden alle Anzeichen einer Atelektase beobachtet. Sie können klein sein und sich mit Schwerpunkten des Emphysems abwechseln.
In einem schweren klinischen Bild, manchmal mit solchen Veränderungen, Mikroabsessen erscheinen, die oft die bronchiale submukösen Drüsen beeinflussen. Dann beginnt der Blitz des Lungenparenchyms, um die Fortschritte, die durch eine akute Form der Lungenentzündung überwacht wird, die bei Patienten mit schweren, langwierigen Verlauf der Mukoviszidose-Klinik, anfällig für Abszessbildung unterscheidet.
Periodisch können die ersten Manifestationen der pulmonalen Form der zystischen Fibrose manchmal im zweiten oder dritten Lebensjahr und auch zu späteren Zeiten auftreten. Die Natur dieser Manifestationen erlangt alle Formen der anhaltenden und schweren Lungenentzündung.
Ältere Kinder sind eher ein klinisches Bild von langwieriger Bronchitis mit klar definierter Obstruktion zu präsentieren. Veränderungen, die durch den entzündlichen Prozeß verursacht werden, manifestieren sich durch diffuse Bronchitis, die sich oft mit verschiedenen Pneumonien von langwieriger Natur wiederholt und sich dann schnell in chronische Lungenentzündungen verwandelt. Dann allmählich entwickeln Bronchiektase, sowie Pneumosklerose. Interstitielles Gewebe ist betroffen und das ist das Ergebnis einer weit verbreiteten Pneumofibrose. Deshalb sind in beiden Lungen feucht, aber häufiger werden kleine Blasenrasen zugehört.
Extrem ungünstig für den Verlauf der zystischen Fibrose ist die Entwicklung der umfangreichen Pneumosclerose und Bronchiektase gefolgt von der Bildung eines eitrigen Prozesses. Alle Arten von obstruktiven Veränderungen, die bei der zystischen Fibrose beobachtet werden, führen im Laufe der Progression zu erhöhten Symptomen des Emphysems, einer ausgeprägten Verletzung der Sehatmung und der Pathologie der Entwicklung der kleinen Zirkulation.
Parallel zur zugrunde liegenden Erkrankung tritt eine Deformation der Brust auf, die terminalen Phalangen werden zu einer Art Trommelfelle, und Herzinsuffizienz entwickelt sich als pulmonales Herz.
Piopnevmotorax, Pneumothorax und Lungenblutung sind selten Komplikationen der zystischen Fibrose. Und wenn die Krankheit durch die Dauer des Flusses gekennzeichnet ist, dann notieren Nasopharyngeal Läsionen, die Polypen, Adenoide und chronische Tonsillitis gehören. Fast alle Kinder werden mit Sinusitis diagnostiziert, in den klinischen Manifestationen, von denen es eine nasale Stimme, Kopfschmerzen und Sekretion der Sekretion aus den nasalen Passagen gibt.
Nach dem klinischen Bild der Erkrankungen des Darms, sowie der Bauchspeicheldrüse, alle Symptome von Patienten mit zystischer Fibrose der wichtigsten Formen der Krankheit. Aber die Veränderungen, die die Aktivität der Enzyme der Bauchspeicheldrüse reduzieren können, besonders bei Kindern, die auf künstliche Fütterung übertragen werden, können durch unzureichende Aufspaltung von Proteinen und Fetten sowie durch ihre Absorption manifestiert werden. Deshalb entwickeln im Darm verrottende Prozesse, die zur Akkumulation von Gasen und konstantem Blähungen führen. So ist es auch bei der Erstuntersuchung des Patienten möglich, eine zystische Fibrose einzugehen, wobei ein charakteristischer, reichlich vorhandener Hocker mit fäulichem, unangenehmem Geruch zu berücksichtigen ist. Fast 20% der Patienten haben rektalen Prolaps.
Einige der Bauchschmerzen unterscheiden sich durch häufige Schmerzen im Abdomen verschiedener Ätiologie. Sie sind von einem krampfartigen Typ mit Blähungen, Schmerzen in den Muskeln von einem verlängerten paroxysmalen Husten im rechten Hypochondrium, im Bereich der Leber.
Die Lokalisation von Schmerzen im epigastrischen Bereich kann auftreten, weil der Magensaft im Duodenum nicht ausreichend neutralisiert ist. Dies geschieht als Folge einer beeinträchtigten Sekretion der Bauchspeicheldrüse. Deshalb, in einer großen Anzahl von verstorbenen Patienten, die oft negative biochemische Ergebnisse haben, berichtet eine Autopsie eine Zirrhose der Leber des Gallenganges. Bei Patienten mit zystischer Fibrose wird ein erhöhter Appetit festgestellt, aber anhaltende Verdauungsstörungen führen ständig zu Hypotrophie. Manchmal entwickelt sich aber sehr selten hypoproteinämisches Ödem. Die Entwicklung der Hypotrophie wird gleichzeitig durch die Entwicklung von Hypochlorämie, Anorexie und metabolischer Alkalose gefördert.
Zystische Fibrose bei Kindern
Da zystische Fibrose eine der häufigsten erblichen Pathologien für heute ist, ist die Häufigkeit ihres Vorkommens bei Kindern unterschiedlichen Alters sehr hoch. Darüber hinaus wird seine Wahrscheinlichkeit der Entwicklung nicht in irgendeiner Weise durch das Kind gehört zu einem bestimmten Geschlecht betroffen sein. Die Krankheit wählt nicht - es ist ein Junge oder ein Mädchen. Die wichtigste Sache zu verstehen ist, dass zystische Fibrose bei Kindern nur durch genetische Defekte entwickelt. Daher können keine schlechten Gewohnheiten, sozialer Status oder Umweltprobleme eine Wahrscheinlichkeit der Geburt von Kindern mit der Pathologie der zystischen Fibrose sein.
Kinder werden mit dieser Krankheit nur diagnostiziert, wenn sie zwei mutierte Gene haben, die sie von ihren Eltern geerbt haben. Deshalb werden in Familien, in denen das Paar, und beide, Träger des Gens zystische Fibrose, Kinder mit dieser Pathologie geboren werden. Aber diejenigen, die von ihren Eltern nur eine Spezies eines solchen Gens geerbt haben, werden als Träger der zystischen Fibrose betrachtet. Solche Kinder werden absolut gesund geboren.
Kinder haben eine Vielzahl von klinischen Manifestationen der zystischen Fibrose. Einige können Veränderungen in der Atemwege erkennen, vor allem Bronchial- und Lungenerkrankungen, andere - schwere Bauchspeicheldrüsenerkrankungen. Der klinische Verlauf der Krankheit eines Typs, für jedes Kind, kann auf unterschiedliche Weise vorgehen.
Es ist wichtig, sich daran zu erinnern, dass zystische Fibrose eine genetische Pathologie ist, die die geistigen Fähigkeiten eines Kindes nicht beeinträchtigt.
In den ersten Lebensmonaten von vielen Kindern wirkt die Hypotrophie unter Beibehaltung des Appetits und erst nach einiger Zeit im Kot sowie Polyfektien entsteht Fett, das die Eltern des Babys beunruhigt, so dass sie besonderes Augenmerk auf sie legen.
Die Haut des Babys ist ein guter Hinweis auf die Diagnose, da es einen "salzigen" Geschmack hat. Deshalb, mit einer solchen Beschwerde, die Eltern zum ersten Mal konsultieren einen Kinderarzt. Vielleicht wird bei solchen Kindern eine scharfe Form von Hypokaliämie und metabolischer Alkalose durch ausgeprägte Elektrolytstörungen manifestieren.
Bei der zystischen Fibrose wird seit dem Atemsystem der Husten das erste Symptom dieser Erkrankung. Am Anfang wird er sich in Form von "husten" manifestieren, dann allmählich zunehmen und den Charakter eines Hustens wie beim Keuchhusten annehmen. Kinder, Zyanose und Kurzatmigkeit, können diesen paroxysalen Husten begleiten, aber es gibt normalerweise keinen Atemstillstand. Ausgeschiedenes Sputum beim Husten, zu Beginn der Erkrankung, helle Farbe und nicht viskos, aber später wird es mukopurulent und erhält eine starke Viskosität. Wenn es ein grünliches Sputum gab, dann ist dies ein Signal über das Aussehen im Körper eines Kindes von Pseudomonas aeruginosa und seine Infektion mit Sputum. In vielen Fällen wird oft ARVI aufgezeichnet, die sich periodisch wiederholen und langwierige Strömungen haben, sowie einen Husten, der lange andauert.
Im Anfangsstadium der zystischen Fibrose ist das Bild der Erkrankung bei der Auskultation von Kleinkindern manchmal normal. Eine gründliche Untersuchung des Kindes ermöglicht es, eine vergrößerte anteroposteriore Größe des Thorax, eine leichte Kurzatmigkeit mit wenig körperlicher Anstrengung und eine reduzierte Auslenkung der Lunge zu erkennen.
Auf der Seite des Magen-Darm-Traktes bei Kindern des ersten Lebensjahres wird ein häufiger flüssiger Hocker mit grünlicher Farbe aufgedeckt, der anschließend fettig wird. Ein Kind mit zystischer Fibrose hat ein Körpergewicht, das nicht mit gesunden Kollegen übereinstimmt, das heißt, hinter der körperlichen Entwicklung zurückbleibt und später auch im Wachstum. Kinder mit zystischer Fibrose, die visuelle Untersuchung haben, haben eine reduzierte Muskelmasse, ein Bauch, der nach außen wölbt, und ein Prolaps des Mastdarms ist oft ein Symptom dieser Pathologie.
Zusätzlich zu den Hauptformen der zystischen Fibrose, zum Beispiel, 15% der neugeborenen Patienten entwickeln meconium Obstruktion des Darms. Symptome dieser Art von Pathologie können Erbrechen mit Galle, die Unmöglichkeit der Divergenz von Meconium und einem vergrößerten Bauch. Grau-Meconium wird im Dünndarm, im Ileocecal-Ventil gehalten. Die gefährlichste Manifestation dieser Form der zystischen Fibrose ist ihre Komplikation in Form von Meconiumperitonitis.
Die Gesamtheit all dieser pathologischen Prozesse von der Seite der Atmung, der Verdauung und der Schweißdrüsen führt in der Regel zu einer Kinderdystrophie. Hypoproteinämie und Anasarca bei Säuglingen treten in einer Kombination von hämolytischer Anämie auf. Oft haben solche Kinder einen Mangel an fettlöslichen Vitaminen. Zum Beispiel ist die Ursache der Blutungen ein Mangel an Vitamin K, als Folge der Hypoprothrombinämie.
Kinder, die an zystischer Fibrose leiden, zeichnen sich durch einen starken Verlust von Salzen im Schwitzen aus, als Folge von Hitze, häufigen entzündeten Nebenhöhlen, die durch Polyposis und parotide Drüsenbeteiligung kompliziert sind.80% der kranken Kinder haben eine erhöhte Empfindlichkeit gegenüber verschiedenen Arten von Allergenen wie Nahrung, Haushalt und Medizin.
Eine der Komplikationen der zystischen Fibrose bei Kindern ist Zöliakie, aber eine häufige Komplikation ist Cholelithiasis.
Cystic fibrosis Diagnose
Diagnose von zystischer Fibrose ist die Basis von chronischen bronchopulmonalen Prozess, Darmsyndrom, einer positiven Schweißtest und Mukoviszidose Geschwister. Deshalb, um diese Pathologie zu diagnostizieren, genügt es, zwei dieser aufgeführten Merkmale zu haben. Es wurden bereits bestimmte Kriterien für die Diagnose der zystischen Fibrose entwickelt, die zwei Diagnosetypen beinhalten.
Das erste basiert auf den charakteristischen klinischen Symptomen. Dies kann sowohl eine Familiengeschichte der zystischen Fibrose und Neugeborenen-Screening für immunoreaktive Trypsin von einer positiven Natur sein.
Der zweite ist der erhöhte Gehalt an Kaliumchloriden( mehr als 60 mmol / l), die identifizierten zwei Mutationen, ein positives Ergebnis bei der Messung der Differenz der Nasenpotentiale. Die Diagnose ist zuverlässig, auch wenn es ein Kriterium von jeder Art von Diagnose gibt.
Zur Diagnose der zystischen Fibrose Resort auf eine Reihe von Methoden, die ziemlich informativ, aber auch zeitaufwendig sind. Hierzu ist es notwendig, die Konzentration von Natriumchlorid im Schweiß zu bestimmen, um Untersuchungen der Stuhlkoliken durchzuführen, um die DNA-Diagnostik herzustellen, um den Unterschied in den Nasenpotentialen zu messen, um die Aktivität der Pankreaselastase im Kot zu bestimmen.
Derzeit gibt es eine echte Chance, eine vorgeburtliche Untersuchung des Fötus auf das Vorhandensein von zystischer Fibrose im Zusammenhang mit der verfügbaren DNA-Diagnostik durchzuführen. Und es ist notwendig, diese Prüfung zu künftigen Eltern auch vor der Schwangerschaftsplanung durchzuführen, dh die Anwesenheit oder Abwesenheit eines zystischen Fibrosegens im Körper zu bestimmen.
Bisher haben die meisten Familien, die ein krankes Kind mit zystischer Fibrose haben, aber die so eine Art Diagnose wie DNA durchgemacht haben, haben in der Zukunft Kinder schon ganz gesund.
Analyse für zystische Fibrose
Es gibt eine Reihe von Tests, Diagnosen und Tests, die verwendet werden, um zystische Fibrose zu diagnostizieren. In den ersten Monaten des Lebens in Neugeborenen kann neonatale Diagnostik durchführen. Diese Methode besteht darin, die Menge an IRT-Gehalt im Blut eines Kindes zu bestimmen.ИРТ ist ein Enzym der Bauchspeicheldrüse. Wenn ein Kind krank mit zystischer Fibrose ist, dann wird die Analyse auf ИРТ mit einem erhöhten Index sein. Eine solche Analyse erfolgt, wenn es Verdacht auf zystische Fibrose gibt.
Der effektivste diagnostische Test für zystische Fibrose ist ein Schweißtest. Das Standardverfahren ist, dass eine Hautstelle für einen Schweißtest genommen wird, auf dem zuvor Ionophorese mit Pilocarpin durchgeführt wurde. Die Konzentration von NaCL in den Sekreten der Schweißdrüsen sollte 40 mmol / l nicht überschreiten. Ist das Ergebnis der Testprobe höher als 60 mmol / l, so kann es als positiv betrachtet werden. Ein wichtiger Punkt ist die Re-Analyse. Es ist getan, wenn die Antwort positiv, zweifelhaft oder negativ ist, aber das klinische Bild deutet auf die Wahrscheinlichkeit einer zystischen Fibrose hin.
Um endlich zu diagnostizieren, ist es notwendig, mehrere solcher Tests durchzuführen, zwei oder drei und erhalten ein bestätigendes positives Ergebnis.
Aber manchmal sind die Antworten falsch. Dies kann auf eine ungenaue Sammlung von Material und dessen Transport zurückzuführen sein. Eine Probe von einem Neugeborenen oder einem Patienten, der Cloxacillin einnimmt.
Unter den Methoden der Forschung gibt es auch coprologische Methoden. Untersuchen kaproliyu kala Patienten, können Sie erkennen, Steatorii. Dies liegt daran, dass der Duodenum eine geringe Aktivität oder völlige Abwesenheit von Pankreasenzymen aufweist.
Wenn die Brustradiographie die Bronchialdichten nachweisen kann und auch eine Erhöhung der Luftigkeit des Lungengewebes anzeigt. Das Röntgenbild zeigt Anzeichen eines Sturzes der Lungen, vor allem des Oberlappens. Dies ist ein Indikator für zystische Fibrose.
Die Verwendung der Untersuchung der Funktionen der externen Atmung macht es möglich zu bestimmen, wie stark das Atmungssystem betroffen ist. Diese Untersuchung bestimmt, wie die Bronchien auf Bronchodilatatoren reagieren und Patienten identifizieren, für die der Zweck dieser Medikamente vernünftig ist.
Für ältere Kinder misst die informativste Methode zur Diagnose einer Krankheit den Unterschied in den Nasenpotenzialen. Seine Hauptaufgabe ist es, den Hauptfehler zu finden, der die Entwicklung der zystischen Fibrose verursacht. Indikatoren werden aus der Nasenschleimhaut und aus der Haut des Unterarms genommen.
Zwei weitere Arten von Analysen können festgestellt werden - genetisch und pränatal. Die erste ist ziemlich teure Analyse, und die zweite ist DNA-Diagnostik.
Behandlung der zystischen Fibrose
Im Moment ist die Medizin in der vollständigen Heilung der zystischen Fibrose ohnmächtig. Wissenschaftler vieler Länder arbeiten an der Entwicklung von Drogen auf zellulärer Ebene, aber leider kann ein positives Ergebnis bisher nicht erreicht werden.Ärzte können nur hoffen, dass irgendwann diese Krankheit vollständig geheilt wird. Die wichtigste Behandlungsmethode könnte "Gentherapie" sein, die für die Bereitstellung von gesunden Kopien eines mutierten Gens an kranke Zellen verantwortlich wäre. Aber das sind nur Annahmen und versuchen, die erbliche Pathologie mit bekannten Methoden zu behandeln.
Die Grundlage für die Behandlung von zystischer Fibrose ist eine schrittweise Annäherung an diese Krankheit. Am wichtigsten ist, ist es notwendig, eine günstige Umgebung zu schaffen, eine bestimmte Diät mit dem Zusatz von Enzymen zuzuordnen, eine Physiotherapie durchzuführen und dem Patienten eine ausreichende körperliche Aktivität zu verleihen.
Für ein erfolgreiches Therapieergebnis ist es notwendig, in einer hochqualifizierten Apotheke und moralischen Unterstützung von nahen Menschen zu beobachten.
Therapeutischer Ansatz zur Behandlung eines Patienten muss ausschließlich einzeln und kontinuierlich durchgeführt werden. Um dies zu tun, ist es wichtig, den infektiösen Prozess in der Atemwege rechtzeitig zu stoppen. Dann wieder die Atemwege und Clearing-Funktion der Bronchien. Und am Ende der Behandlung, passen Sie den Mangel an Enzymen in der Bauchspeicheldrüse. Prozess
Therapie von Lungenerkrankungen umfasst diejenigen Ereignisse, die Sputum Viskosität verringern und bronchiale Entwässerungs- und antimikrobielle Therapie verbessern, um die Behandlung einer Vergiftung, Hypoxie, Vitaminmangel und Herzinsuffizienz.
Um die Viskosität von Sputum zu reduzieren, sind enzymatische Präparate wie Hymopsin, Chymotrypsin, Fibrinolysin oder mucoletische Medikamente vorgeschrieben. Tabletten Mukosolvin aufgenommen und intramuskulär - Acetylcythetein. Sie können sowohl Bromhexin als auch Muciltin verwenden, aber sie haben eine schwache Verdünnungseffekt. Für eine gute Drainage der Bronchien ist eine Massage des Thoraxbereichs der Lunge und der therapeutischen Gymnastik notwendig. Säuglinge, mit einer elektrischen Pumpe, produzieren Sputum. Jeder Patient sollte einen Kompressor-Inhalator haben, mit dem es möglich ist, ein wirksames Medikament Pulmozim einzuführen.
Wenn Lungenkrankheiten in den Prozess der Exazerbation sind, dann Rückgriff auf die Ernennung von Antibiotika-Therapie-Kurs für 3-4 Wochen. Um dies zu tun, müssen sie ein Antibiotikum machen, aber wenn es unmöglich ist zu bestimmen, dann für die Zwecke der Medikamente sind Pathogene zystische Fibrose - Pseudomonas aeruginosa und Staphylokokken.
Gleichzeitig mit antibakteriellen Medikamenten sind Antihistaminika und antimikrobielle Mittel vorgeschrieben. Zum Zeitpunkt der Verschärfung der Erkrankung werden UHF-Therapie und Magnesium-Elektrophorese durchgeführt. Um die Hypertonie der Lunge zu reduzieren, ist Euphyllin in Tabletten mit 7-10 mg / kg pro Tag vorgeschrieben. Um den Stoffwechsel des Myokards zu verbessern, werden die Ziele von Cocarboxylase und Kalium-Orotat gezeigt. Mit einem pulmonalen Herzen nehmen sie Digoxin, Glucocorticotiden( 1-1,5 mg / kg pro Tag).
die Behandlung von Darmsyndrom Patienten verschrieben neue Enzympräparate beschichtet und resistent gegen die saure Umgebung - es pantsitrat und Kreon, der effektiv Mezim forte, festal oder panzinorm. Bei Patienten mit zystischer Fibrose müssen diese Medikamente im Laufe des Lebens eingenommen werden, und die Dosierung ist für jeden Patienten streng individuell vorgeschrieben. Angemessene Schmerzen im Bauch, ein normalisierter Stuhl und ein Mangel an neutralem Fett bei der Untersuchung von Fäkalien für die Scatologie gelten als ausreichend für das Enzym genommen werden.
Unabhängig von dem verallgemeinerten Prozess und den Veränderungen, die in vielen Organen und Systemen auftreten, ist die Abgabe von kranken Kindern durch einen Bezirksarzt und einen Pulmonologen obligatorisch. Darüber hinaus sollten die Eltern lernen, wie man sich um ein krankes Kind kümmert, ihre eigenen Massagen, therapeutische Gymnastik und Aerosoltherapie durchführt. Klinische Überwachung ist notwendig, um die Funktionen der Arbeit der Bronchien, Lunge, Herz, Magen, Darm, Leber, Nieren und den korrekten Empfang von Enzympräparaten für die rechtzeitige medizinische Versorgung bei akuten Exazerbationen der Krankheit zu kontrollieren, für die Therapie zu stärken, sowie für die obligatorische Linderung von chronischen Infektionen.
Die meisten Kinder mit zystischer Fibrose sind ambulant, wo das Kind mit häuslicher Pflege voll versorgt wird und der Rückfall von Infektionen ausgeschlossen werden kann. Nur bei schweren Formen der Krankheit sind in der Regel Krankenhaus-Patienten. Dies bezieht sich auf Atemversagen von II-III Grad, Dekompensation des pulmonalen Herzens und Hämoptyse. Meconial Ileus, sowie Darm-Obstruktion, in denen konservative Behandlung ist nicht wirksam, ist ein Hinweis für chirurgische Eingriffe.
Es kann auch festgestellt werden, dass es radikalere Methoden zur Behandlung von zystischer Fibrose gibt, wo sie die Organtransplantation nach Vitalzeichen betrachten. Dies wird vor allem in Kliniken in Deutschland, Kanada und den USA praktiziert. Es sind diese Operationen, die einigen Patienten mit erblicher Pathologie helfen, zu überleben und lange genug zu leben.
Es gibt natürlich unkonventionelle Behandlungsmethoden. Aber sie sind mehr Hilfsmittel und werden vor dem Hintergrund des Hauptes angewendet.
Kinder, die leichte bis mäßige Darmzystenfibrose haben, können in einem Sanatorium behandelt werden. Wenn es möglich ist, spezielle Gruppen für pulmonale Formen der Krankheit zu schaffen, ist es auch nützlich für solche Kinder, Sanatorium Behandlung zu nehmen. Die Kriterien für die Auswahl der Patienten für diese Art der Behandlung sind nach der Einnahme der Enzympräparat Darm-Störungen kompensieren, die keine Fehlfunktionen von Lungen-Herz-und alle Arten von Entzündungen sind.
Es ist nicht empfehlenswert für kranke Kinder mit zystischer Fibrose in Kindergärten und Krippe zu sein. Aber du kannst nur in guter oder befriedigender Schule zur Schule gehen, aber du musst noch einen zusätzlichen Tag unter der Woche haben. Während der Untersuchung oder Behandlung sind diese Kinder von der Teilnahme am Schulunterricht befreit und alle Prüfungen bestanden.
Alle Kinder mit zystischer Fibrose diagnostiziert sind auf eine Gesundheits-Check-up für das Leben. Das einzige ist, dass sie im Alter von 15 Jahren auf das Beobachtungskonto übertragen werden, an den Therapeuten.