Prader-Willi sündroom
 Prader-Willi sündroom - geneetilise haruldane anomaalia iseloomustab toimingu lõpetamist geenide või teaduslikus mõttes on see nähtus on tingitud ekspressiooni puudumine, mis tähendab üleandmise geneetiline informatsioon DNA-lt RNA.Sellistel juhtudel tuleb geenide funktsioon kaasata töö õigeaegselt. Näiteks poisid ei ole lapseeas näo juuste kasvu ja see nähtus algab pärast puberteedi algust. Seda haigust kirjeldati esmakordselt 1956. aastal.
Prader-Willi sündroom - geneetilise haruldane anomaalia iseloomustab toimingu lõpetamist geenide või teaduslikus mõttes on see nähtus on tingitud ekspressiooni puudumine, mis tähendab üleandmise geneetiline informatsioon DNA-lt RNA.Sellistel juhtudel tuleb geenide funktsioon kaasata töö õigeaegselt. Näiteks poisid ei ole lapseeas näo juuste kasvu ja see nähtus algab pärast puberteedi algust. Seda haigust kirjeldati esmakordselt 1956. aastal.
Prader-Willi sündroom esineb sagedusega ühel juhul 12000 sündinud lapse kohta. Seda haigust kirjeldasid tema teosed Heinrich Willy, Andrea Prader, Andrew Ziegler, Alexis Labhart, Guido Fanzoni.
Pradera-Willy sündroom
põhjused Seda haigust iseloomustab isa päritud viieteistkümnenda kromosoomi seitsme geeni puudumine või ekspresseerimine. Prader-Willi sündroomi on täheldatud muutustega isa kromosoomis ja ema kromosoomi muutuse korral on täheldatud Angelmanni sündroomi.
Prader-Willi sündroomi tekkega geenide komplektis on aktiivselt aktiivne isendilt saadud geeni koopia ja ema seda ei tee. See tähendab, et kui enamikul inimestel on nende geenide üks koopia, siis Prader-Willi sündroomiga patsiendid elavad ilma sellise koopia.
Prader-Willi sündroomi sümptomite
selle haiguse iseloomustab puusaliigese düsplaasia, hüpotoonia( madal lihastoonus), rasvumine( tausta liigsöömist tekivad 2. aasta).Kannatavad Prader-Willi sündroom on vähendatud liigutuste koordinatsiooni, samuti madal luutihedus, on omanikud väikeste käte ja jalgade, lühikest kasvu, valmis magama, STRABISM, on selgroo kõverus. Sellistele inimestele iseloomustab paks sülg, hüpogonadism( suguelundite funktsiooni langus), halvad hambad, viljatus. Patsiente iseloomustab vaimne alaareng, samuti kõne areng, puberteedi mahajäämus ja motoorsete oskuste omandamise raskused.
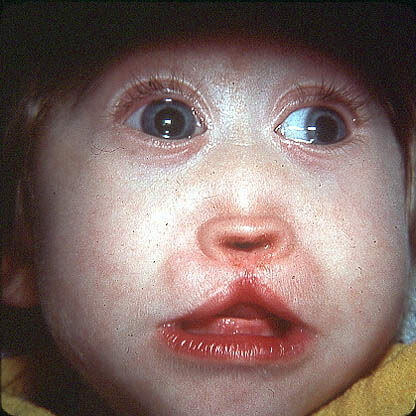
Prader-Willi sündroom ja selle haiguse sümptomid on visuaalselt tähistatud suure nina silla, kitsa ja kõrge esiotsaga, mandlikujulised silmad, kitsad huuled. Kõik need nähud ja sümptomid ei ilmne ühel patsiendil. Sageli vastab patsient kuni viiest ülaltoodud sümptomist.
Prader-Willi sündroomi diagnoos
Seda haigust saab diagnoosida enne sündi. Selle põhjuseks on loote madal liikuvus või sageli lapse vale positsioon, samuti mitmehüdrimaskused( amniokene vedelik ülemäärases koguses).
Prader-Willi sündroomi diagnoositakse sageli pärast geneetilist analüüsi. Seda analüüsi teeb vastsündinu, kellel on hüpotooniline( vähenenud lihastoonus).Mõnikord teevad arstid vigu ja diagnoosivad Downi sündroomi või müopaatiat. Märgid diagnoosimise pärast sündi hõlmavad: sünnist kuni keisrilõike Lukk esitlus, hüpotensioon, letargia, nõrk imemiseks refleks, sest nõrgenenud lihastoonust väikelapse, hingamisraskused, hüpogonadismi.
Prader-Willi sündroomiga lapsed on sarnased, nii kogenud geneetiline diagnoosib viivitamatult haigust, isegi ilma vereanalüüsi tulemusi oodates.
Prader-Willi sündroom ja Angelmanni sündroom
Praeguseks on tuntud veel üks Prader Willy sündroomi tuntud haigus. Seda haigust nimetati Angelmaniks. Seda iseloomustab emaka geneetilise materjali mutatsioon. Enamiku iseseisvate uuringute tulemused viitavad haiguse põhjustele kui mutatsioonile geenis. Selle geeni produkt on kogu komplekssüsteemi proteiini lagunemise ensüümkomponent. See haigus on nime saanud Suurbritannia lastearst, Harry Angelmann, kes kirjeldas teda 1965. aastal.
Prader-Willi sündroomi ravi
Haigus, mis on kaasasündinud geneetiline kõrvalekalle, jääb ebaselgeks ja selle ravimeetodid pole välja töötatud.
Kuidas Prader-Willi sündroomi ravida? Siiski on mõned meditsiinilised meetmed, mis parandavad inimeste elukvaliteeti. Näiteks lastel, kellel on hüpotensioon, on vaja massaaži, samuti muid eriteraapiaseireid. See näitab spetsiifiliste meetodite kasutamist lapse arengu parandamiseks, samuti defektioloogi ja logopeediga klasside kasutamist. Prader-Willi sündroomiga 7, 8, 9-aastastel lastel on määratud kasvuhormoonid, samuti on näidustatud hormonaalne ravi gonadotropiinidega.
Prader-Willy sündroom poistel avaldub hüpogonadismil, mikropüaiaga, samuti krüptoršidismil( mitte munandite ovulatsioon).Sellises olukorras soovitavad arstid või oodake, kuni munandid hakkavad langema, või soovitame operatsiooni hormoonraviga. Kaaluvõtu parandamiseks on ette nähtud dieet, mis piirab süsivesikute ja rasvade hulka. Kui rasvumine on juba olemas, tuleb jälgida Prader-Willi sündroomiga patsientide imendunud toidu kogust ja kvaliteeti. Selliste inimeste jaoks on iseloomulik huntlik isu. Apnea haiguse kulgu võib tekkida komplikatsioon, mida iseloomustab hingamise hilinemine unenägudes.
Prader-Willie sündroomi prognoos
Teise lapse planeerimisel võivad samadel vanematel olla teine haigus. See sõltub geneetilisest rikeest põhjustatud mehhanismist. Risk on väiksem kui 1% juhtudel, kui esimesel lapsel on geeni kustutamine või ühe vanemaga ostehenogeneetiline disosmia. Risk on kuni 50% tingimusel, et rike on tingitud mutatsioonist. Kuni 25% risk on vanemate kromosoomide translokatsioon. Igal juhul peavad vanemad tegema geneetilist eksami.
Prader-Willi sündroom ja selle prognoos hoiavad sageli kõne ja vaimse arengu viivitust. Freim ja Kerfsi uuringud näitasid, et 5% patsientidest on keskmine luurekoefitsient. See võrdub IQ skaalal 85 või enama punktiga.27% patsientidest on keskmise äärega luureandmete tase, mis on hinnanguliselt 70-85 punkti.34% -l patsientidest on luuretase 50-70 punkti.27% patsientidest jäävad keskmise lõhe tasemele ja on hinnanguliselt 35-70 punkti.5% -l patsientidest on tugev lag ja nad saavad 20-35 punkti ja vähem kui 1% -l on märkimisväärne lag. Teistes uuringutes on andmeid, et 40% patsientidest näitavad keskmist või vähenenud luureandmeid.
Prader-Willi sündroomiga lastel on tavaliselt hea pikaajaline ja visuaalne mälu. Nad suudavad õppida lugema, neil on passiivne, rikas sõnavara, kuid nende enda kõne on tihti hullem kui tema arusaam. Kuulmismälu, kirjutamisoskus ja matemaatilised oskused, lühiajaline kuulmis- ja visuaalne mälu kannatavad ka ja ei vasta vanusepiirangutele.
Prader-Willi sündroomi põhjustab sageli suurenenud söögiisu, kuna patsiendid on suurendanud griinhormooni sisaldust veres. Patsientide puhul on somatoliberiini madal kontsentratsioon tüüpiline. Mõned seostavad seda 15-nda kromosoomiga, mis on seotud hüpotalamusega. Teised andmed näitavad hüpotalamuse paraventrikulaarsete tuumade arvu vähenemist nii rakkude kui ka oksütotsiini sisaldavate rakkude koguarvust. Kuid huvitavalt on tõendeid, mis viitavad vastupidi: surnute lahkamine selle haigusega ei näita sageli hüpotalamuse vigu.