Malattia di Gaucher
 La malattia di Gaucher è una malattia accumulata ereditaria che è uno dei più comuni tra le patologie lisosomiali.malattia di Gaucher sviluppa come risultato della carenza di glucocerebrosidasi, che diventa il risultato dell'accumulo di glucocerebroside nei tessuti e alcuni organi.
La malattia di Gaucher è una malattia accumulata ereditaria che è uno dei più comuni tra le patologie lisosomiali.malattia di Gaucher sviluppa come risultato della carenza di glucocerebrosidasi, che diventa il risultato dell'accumulo di glucocerebroside nei tessuti e alcuni organi.
Questa malattia è una malattia relativamente rara e si manifesta, secondo le statistiche, relativamente basse. Tuttavia, tuttavia, può portare alla disabilità e / o alla morte in età precoce, se il trattamento tempestivo non è stato avviato.la malattia di Gaucher
provoca
a livello genetico, le mutazioni si verificano in geni che sono responsabili per la produzione dell'enzima glucocerebrosidasi. Questo gene con anomalie è localizzato sul primo cromosoma. Queste mutazioni provocano una bassa attività dell'enzima. Pertanto, vi è un accumulo di glucocerebrosidi nei macrofagi.
Le cellule mesenchimali, note come le cellule di Gaucher, si espandono gradualmente e diventano ipertroficate. Poiché queste cellule sono derivate modificazioni, e sono nella milza, rene, fegato, polmone, cervello e del midollo osseo, che, a sua volta, distorcono questi corpi e interrompono il loro normale funzionamento.
La malattia di Gaucher si riferisce a malattie autosomiche recessive. Pertanto, ogni persona può ereditare una mutazione di questo enzima con tutte le caratteristiche dello stesso rapporto, sia dal padre che dalla madre. Quindi, il grado della malattia e la sua gravità dipenderanno dalla sconfitta dei geni.
Teoricamente, ogni persona può ereditare un gene glucocerebroside con lesioni o assolutamente sani. Come conseguenza dell'eredità di un gene con anomalie, si verifica una mutazione di questo enzima, ma questo non parla della malattia. Ma quando il bambino riceve entrambi i geni affetti, viene diagnosticata la malattia di Gaucher. Se uno dei geni colpiti è ereditato, il bambino è considerato il vettore della malattia, quindi c'è una probabilità di trasmissione di questo tratto, con patologia ereditaria, per le generazioni future. Così, con il trasporto della malattia da entrambi i genitori, il bambino può nascere con la malattia di Gaucher nel 25% dei casi, il bambino portatore nel 50% e il bambino sano nel 25%.
L'incidenza di questa patologia ereditaria tra le razze etniche è di 1: 50.000, ma è più spesso presente tra gli ebrei Ashkenazi.malattia di Gaucher
è chiamato anche malattie da accumulo da carenza dell'enzima, che consiste nel visualizzare il corpo di prodotti metabolici nocivi e non accumulare loro. Di conseguenza, queste sostanze si raccolgono nei macrofagi di alcuni organi e li distruggono.
Malattia di Gaucher Sintomi
L'immagine clinica della malattia è caratterizzata da tre tipi.
Nel primo tipo di malattia di Gaucher sistema nervoso è coinvolto nella accumulo di lipidi, quindi si riferisce più alla malattia ematologica flusso. In primo luogo, c'è un aumento di molti organi parenchimali, il primo posto tra cui è la milza, e poi sono osservate deformazioni visive dalle ossa. Molto più spesso questo tipo di malattia progredisce tra gli adulti.
L'infanzia o il secondo tipo di malattia di Gaucher è caratterizzata da una forma acuta della neurologia e viene diagnosticata, soprattutto, fino a sei mesi di età.In questo caso, una comparsa molto precoce di caratteristici sintomi neurologici, in cui le cellule staminali del cervello sono danneggiate, causando la morte nei bambini.
Il terzo tipo di malattia di Gaucher è una forma giovanile della malattia con un tipo neuroviscerale subacuto. All'inizio del processo patologico si sviluppa splenomegalia e ritardo mentale, in cui sono interessati sistemi cerebrali piramidali e extrapiramidali.
La malattia di Gaucher ha grandi cambiamenti in forma di danno al midollo osseo, alla milza, ai linfonodi e al fegato, mentre la forma cerebrale è influenzata anche dal cervello. Negli organi parenchimali c'è un ampio contenuto di cellule Gaucher e, con le aree esistenti di necrosi, si verificano cambiamenti diffusi nella corteccia cerebrale. Durante l'esame dei pazienti, la marcatura della milza e del fegato aumentava notevolmente, e talvolta i linfonodi. Sul corpo appare caratteristica per questo macchie di malattia di colore giallo scuro.
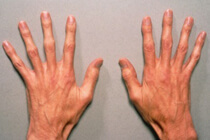
La malattia di Gaucher presenta un sintomo distintivo nella clinica della malattia sotto forma di un aumento della milza all'inizio del patologie in via di sviluppo, e poi c'è un aumento delle dimensioni e del fegato. Si osserva una certa pigmentazione sul viso, le mani, le sclere e le mucose. E l'accumulo di lipidi nel midollo osseo porta ad osteoporosi delle vertebre, femori, fratture spontanee, anemia, trombocitopenia e leucopenia. A volte i neonati hanno anche una forma maligna della malattia, che si verifica sotto forma di sindrome spastica, strabismo, tosse come la pertosse.
Nei casi acuti della malattia di Gaucher viene rilevata l'epatosplenomegalia con linfonodi viscerali allargati. Le parti aperte del corpo sono pigmentate con un colore marrone giallastro, che diventa poi bronzo a causa della deposizione di un certo pigmento sotto la pelle. I pazienti, di regola, si lamentano di dolore nelle ossa, deformità caratteristiche e fratture frequenti delle ossa. Nell'età avanzata, ci sono ben definiti sintomi di atheroma vascolare contemporaneamente con aumento della colesterolemia.
Nell'ultima fase della malattia di Gaucher si sviluppano spesso forti forme di disturbo neurologico, che portano alla morte.
Diagnosi della malattia di Gaucher
Oggi diversi metodi sono usati per diagnosticare la malattia di Gaucher.
Uno dei più precisi è un test di sangue, in cui viene determinata la presenza di un enzima e il livello di glucocerebrosidasi nei leucociti.
Nell'analisi del DNA, sono considerati il secondo metodo, le mutazioni a livello genetico e la quantità mancante dell'enzima di base. Questo metodo si basa sulle ultime tecnologie di biologia molecolare. Il principale vantaggio di questa diagnosi di malattia di Gaucher è che ti permette di effettuare esami in gravidanza. E questo consente al 90%, in futuro, di determinare il vettore della malattia e persino la sua gravità.
Nel terzo metodo di diagnosi viene esaminato il midollo osseo, che consente di determinare la disfunzione delle sue cellule. Il lato negativo di questo sondaggio è che permette di diagnosticare solo i pazienti, ma non può identificare i portatori della malattia. Pertanto per oggi tale metodo non viene praticamente applicato.
Purtroppo, a volte i sintomi della malattia di Gaucher non sono sempre facili. Pertanto, la diagnosi corretta e tempestiva può prolungare la vita del paziente per molti anni. In questo caso, il paziente sarà sotto la supervisione di specialisti( pediatri, ematologi), senza ricorrere all'uso del trattamento farmacologico e senza timore di complicazioni. E, al contrario, se un paziente che non abbia superato una diagnosi qualificata in modo tempestivo e non abbia ricevuto assistenza medica, possa avere sintomi della malattia durante tutta la sua vita, soffrirne da loro e non credere che abbia malattia di Gaucher, rispettivamente, e non abbia il trattamento giusto.
trattamento della malattia di Gaucher
Fino a poco tempo fa la malattia è stata trattata in base ai suoi sintomi: la milza è stata rimossa o sono state eseguite operazioni in fratture patologiche. Ma nel 1991, la prima preparazione medica Aglucerase è apparsa negli Stati Uniti, che consente di trattare la malattia di Gaucher. Questa malattia è stata la prima tra le malattie di accumulo, suscettibili alla terapia con sostituti enzimatici.
Il secondo prodotto della terapia sostitutiva dell'enzima per la malattia di Gaucher, imiglucerasi, è stata ufficialmente adottata nel 1994.Questi farmaci sono analoghi dell'enzima umana di glucocerebrosidasi, che vengono prodotti usando le ultime tecnologie di creazione artificiale del DNA.
Al momento, un gran numero di pazienti con malattia di Gaucher in tutto il mondo riceve la terapia sostitutiva fermentativa sotto forma di forme modificate di beta-glucocerebrosidasi. Questi includono ceradasi( alglucerasi) o ceresime( imiglucerasi in iniezioni).Questi farmaci sono progettati in modo tale da poter specificamente targetare i globuli bianchi( macrofagi) che distruggono le infezioni, per accelerare il processo di suddivisione delle glucocerebrosidi in glucosio e ceramide.
Documentato il successo del trattamento della malattia di Gaucher con l'uso di cerevisia ad una dose iniziale di 60 unità per kg, applicata ogni due settimane. Dopo aver completato il corso, si registra che questo particolare dosaggio riduce notevolmente l'organomegalia, riduce spesso gli organi interni nelle dimensioni e le complicazioni dell'etologia etiologica e migliora anche la vita dei pazienti con il primo tipo di malattia di Gaucher.
Dal 1997, questa eredità patologica è stata trattata con la terapia sostitutiva dell'enzima in Russia. Per i pazienti del primo tipo, l'appuntamento di Cerezima inizia con 30 unità al kg una volta al giorno. Sei mesi dopo l'inizio del corso di trattamento per la malattia di Gaucher, sono note dinamiche positive in molti organi parenchimali, così come nei parametri ematologici.
Il trattamento a lungo termine della malattia di Gaucher con questo farmaco stabilizza completamente il processo patologico, riduce notevoli cambiamenti nelle ossa e migliora significativamente la vita dei pazienti. Pertanto, prima di iniziare la terapia corrispondente, i risultati saranno più efficaci.
Un punto importante è la prevenzione della malattia di Gauchers, che consiste nel condurre consulenza genetica medica per le famiglie e diagnosi prenatale. Ciò consente di determinare l'attività della glucocerebrosidasi nelle fasi iniziali della busta fetale e le cellule del sangue del suo cordone ombelicale.