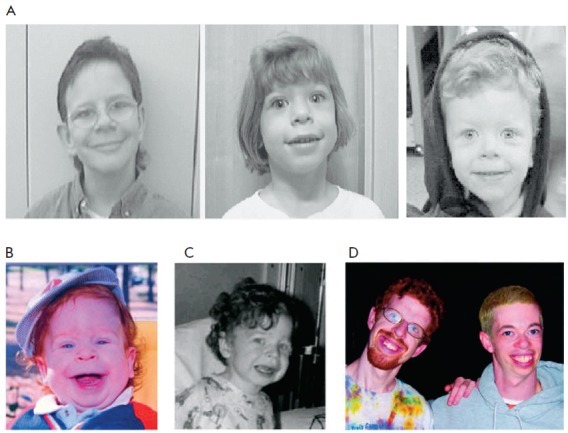
Lipoidoza (lizosomalna choroba spichrzania lipidów): co to jest, przykłady
Uwaga! Informacje służą wyłącznie celom informacyjnym i nie mają na celu diagnozowania i przepisywania leczenia. Zawsze skonsultuj się z lekarzem specjalistą!
Zadowolony
- Czym są choroby spichrzania lizosomalnego?
- Czym są lipidy?
- Jak dziedziczy się lipoidozę?
- Rodzaje i objawy chorób spichrzania lizosomalnego
Czym są choroby spichrzania lizosomalnego?
Choroby spichrzania lizosomalnego (lipoidoza), to grupa dziedzicznych chorób metabolicznych, w których w różnych komórkach i tkankach organizmu gromadzą się szkodliwe ilości substancji tłuszczowych (lipidów).
Osoby z tymi zaburzeniami albo nie wytwarzają wystarczającej ilości jednego z enzymów potrzebnych do rozkładu (metabolizacji) lipidów, albo wytwarzają enzymy, które nie działają prawidłowo.
Z czasem ta nadmierna akumulacja tłuszczu może prowadzić do trwałego uszkodzenia komórek i tkanek, szczególnie w mózgu, obwodowym układzie nerwowym (nerwy od rdzenia kręgowego do reszty) ciało), wątroba, śledziona i szpik kostny.
Czym są lipidy?
Lipidy są substancjami podobnymi do tłuszczu, które są ważnymi częściami błon wewnątrz i między komórkami oraz w osłonce mielinowej, która pokrywa i chroni nerwy. Lipidy obejmują oleje, kwasy tłuszczowe, woski, sterydy (takie jak cholesterol i estrogen) i inne pokrewne związki.
Te substancje tłuszczowe są naturalnie magazynowane w komórkach, narządach i tkankach organizmu. Małe ciałka w komórkach, zwane lizosomami, regularnie przekształcają lub metabolizują lipidy i białka w mniejsze składniki, aby dostarczyć organizmowi energię. Choroby, w których materiał wewnątrzkomórkowy, który nie może być metabolizowany, pozostaje w lizosomach, nazywane są chorobami spichrzania lizosomalnego.
Oprócz chorób związanych z akumulacją lipidów, inne choroby lizosomalne związane z akumulacją obejmują mukolipidozy, w których gromadzą się komórki i tkanki nadmierna ilość lipidów z przyczepionymi do nich cząsteczkami cukru oraz mukopolisacharydoz, w których gromadzi się nadmierna ilość dużego, złożonego cukru Cząsteczki.
Jak są dziedziczone lipoidoza?
Choroby spichrzania lizosomalnego są dziedziczone od jednego lub obojga rodziców, którzy są nosicielami wadliwego genu regulującego działanie określonego enzymu metabolizującego lipidy w klasie komórek organizmu. Mogą być dziedziczone na dwa sposoby:
- Dziedziczenie autosomalne recesywne występuje, gdy oboje rodzice niosą i przekazują kopię wadliwego genu, ale żaden z rodziców nie jest dotknięty chorobą. Każde dziecko urodzone przez tych rodziców ma 25% szans na odziedziczenie obu kopii wadliwego genu, 50% prawdopodobieństwo, że będzie nosicielem podobnym do swoich rodziców i 25 procent szans na nieodziedziczenie żadnej kopii wadliwego gen. Dzieci obu płci mogą być dziedziczone w sposób autosomalny recesywny.
- Dziedziczenie recesywne sprzężone z chromosomem X (lub związane z płcią) występuje, gdy matka jest nosicielem dotkniętego genu na chromosomie X. Chromosomy X i Y biorą udział w determinacji płci. Kobiety mają dwa chromosomy X, podczas gdy mężczyźni mają jeden chromosom X i jeden chromosom Y. Synowie nosicielek mają 50 procent szans na odziedziczenie i zarażenie tej choroby, ponieważ synowie otrzymują jeden chromosom X od matki i jeden chromosom Y od ojca. Córki mają 50 procent szans na odziedziczenie dotkniętego chromosomu X od matki i są nosicielkami lub są w niewielkim stopniu dotknięte chorobą. Dotknięci mężczyźni nie przenoszą choroby na swoich synów, ale ich córki będą nosicielami i nosicielkami choroby.
Rodzaje i objawy chorób spichrzania lizosomalnego
choroba Gauchera - najczęstsza z chorób lizosomalnych. Choroba jest spowodowana niedoborem enzymu glukocerebrozydazy, który prowadzi do akumulacji glukocerebrozydu w wielu tkankach. Tłuszcz może gromadzić się w mózgu, śledzionie, wątrobie, nerkach, płucach i szpiku kostnym.
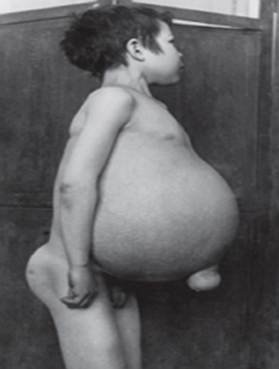
Objawy mogą obejmować uszkodzenie mózgu, powiększenie śledziony i wątroby, zaburzenia czynności wątroby, choroby układu kostnego i uszkodzenia kości, które mogą powodować ból i złamania, obrzęk węzłów chłonnych i (czasami) sąsiednich stawów, wzdęciabrązowawy odcień skóry, niedokrwistość, mała liczba płytek krwi i żółte plamki na oczach.
Osoby najbardziej dotknięte chorobą mogą być również bardziej podatne na infekcje. Choroba dotyka w równym stopniu mężczyzn, jak i kobiety.
Choroba Gauchera ma trzy ogólne podtypy kliniczne:
- Typ 1 (Albo nie neuropatia typ) jest najczęstszą postacią choroby w Stanach Zjednoczonych i Europie. Mózg nie jest dotknięty, ale może dojść do uszkodzenia płuc i, w rzadkich przypadkach, nerek. Objawy mogą rozpocząć się we wczesnym okresie życia lub w wieku dorosłym i obejmują powiększenie wątroby i poważne powiększenie śledziony, co może prowadzić do pęknięcia i dodatkowych powikłań. Osłabienie szkieletu i choroba kości mogą być rozległe. Osoby z tej grupy zwykle łatwo ulegają siniakom ze względu na niską liczbę płytek krwi we krwi. Mogą również odczuwać zmęczenie z powodu anemii. W zależności od początku i ciężkości choroby, osoby z typ 1 może dożyć dorosłości. Wiele osób cierpi na łagodną chorobę lub może nie wykazywać żadnych objawów. Pomimo 1rodzaj Choroba Gauchera jest powszechna wśród osób pochodzenia żydowskiego aszkenazyjskiego i może dotykać ludzi o różnym pochodzeniu etnicznym.
- Wpisz 2 (lub ostra neuropatia dziecięca choroba Gauchera) zwykle rozpoczyna się w ciągu 3 miesięcy od urodzenia. Objawy obejmują rozległe i postępujące uszkodzenie mózgu, spastyczność, drgawki, sztywność kończyn, powiększenie wątroby (hepatomegalia wątroby) i śledziony (splenomegalia), nieprawidłowe ruchy gałek ocznych oraz słaba zdolność ssania i połykania. Dzieci dotknięte chorobą zwykle umierają przed ukończeniem 2 roku życia.
- Wpisz 3 (przewlekła neuropatia forma) może rozpocząć się w dowolnym momencie w dzieciństwie, a nawet w wieku dorosłym. Charakteryzuje się powoli postępującymi, ale mniej poważnymi objawami neurologicznymi w porównaniu z ostrą chorobą Gauchera 2 rodzaje. Główne objawy to zaburzenia ruchu gałek ocznych, deficyty poznawcze, słaba koordynacja, drgawki, powiększenie śledziony i/lub wątroby, zaburzenia układu kostnego, zaburzenia krwi, w tym anemia oraz problemy z oddechowy. Prawie wszyscy z chorobą Gauchera 3 rodzaje, kto otrzyma enzymatyczną terapię zastępczą, osiągnie dorosłość.
Dla pacjentów I i III typ Enzymatyczna terapia zastępcza, podawana dożylnie co dwa tygodnie, może radykalnie zmniejszyć wielkość wątroby i śledziony, zmniejszyć nieprawidłowości szkieletu i odwrócić inne objawy. Udany przeszczep szpiku kostnego leczy neurologiczne objawy choroby. Jednak ta procedura niesie ze sobą znaczne ryzyko i jest rzadko wykonywana u osób z chorobą Gauchera.
W rzadkich przypadkach może być wymagany zabieg chirurgiczny w celu usunięcia całości lub części śledziony (jeśli dana osoba ma bardzo niską liczbę płytek krwi lub gdy powiększony narząd znacznie wpływa na komfort osoby). Transfuzja krwi może przynieść korzyści niektórym osobom z anemią. Inni mogą potrzebować operacji wymiany stawu, aby poprawić mobilność i jakość życia. Obecnie nie ma skutecznego leczenia uszkodzenia mózgu, które może wystąpić u osób z chorobą Gauchera typu 2 i 3.
choroba Niemanna-Picka to grupa zaburzeń autosomalnych recesywnych spowodowanych gromadzeniem się tłuszczu i cholesterolu w komórkach wątroby, śledziony, szpiku kostnego, płuc i, w niektórych przypadkach, mózgu.
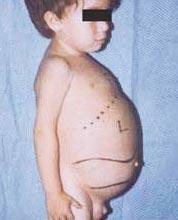
Powikłania neurologiczne mogą obejmować ataksję (brak koordynacji mięśniowej, która może wpływać na chodzenie, pisanie i jedzenie, między innymi), paraliż oczu, zwyrodnienie mózgu, problemy z nauką, spastyczność, trudności w karmieniu i połykaniu, niewyraźne mowa, utrata napięcia mięśniowego, nadwrażliwość na dotyk i pewne zmętnienie rogówki z powodu nadmiernego nagromadzenia materiały. U 50% chorych wokół środka siatkówki pojawia się charakterystyczna wiśniowo-czerwona aureola, którą lekarz może zaobserwować za pomocą specjalnego przyrządu.
Choroba Niemanna-Picka dzieli się na trzy ogólne podtypy:
- Typ A, najcięższa forma, zaczyna się we wczesnym dzieciństwie. Niemowlęta po urodzeniu wyglądają normalnie, ale po 6 miesiącach rozwijają się głębokie uszkodzenia mózgu, powiększona wątroba i śledziona oraz powiększone węzły chłonne i węzły pod skórą (Xanthomas). Śledziona może stać się 10 razy większa od normalnego i może pęknąć, powodując krwawienie. Dzieci te stają się coraz słabsze, tracą funkcje motoryczne, mogą stać się anemiczne i podatne na nawracające infekcje. Rzadko żyją dłużej niż 18 miesięcy. Ta postać choroby występuje najczęściej w rodzinach żydowskich.
- Typ B(lub młodzieńczy początek) zwykle nie wpływa na mózg, ale większość dzieci rozwija się ataksja, uszkodzenie nerwów opuszczających rdzeń kręgowy (Neuropatia obwodowa) oraz trudności płucne, które postępują wraz z wiekiem. Powiększenie wątroby i śledziony jest typowe dla młodzieży. Ludzie z typ B może żyć stosunkowo długo, ale wiele z nich wymaga dożywotniej tlenoterapii z powodu uszkodzenia płuc. Typy A i B Niemanna-Picka są wynikiem nagromadzenia się substancji tłuszczowej zwanej sfingomielina z powodu niedoboru enzymu zwanego sfingomielinazą.
- Typ C może pojawić się we wczesnym okresie życia lub rozwinąć się w okresie dojrzewania, a nawet w wieku dorosłym. choroba Niemanna-Pickatyp C nie jest spowodowane niedoborem sfingomielinazy, ale brakiem białek NPC1 lub NPC2. W rezultacie różne lipidy, a zwłaszcza cholesterol, gromadzą się w komórkach nerwowych i powodują ich nieprawidłowe działanie. Zaangażowanie mózgu może być rozległe, powodując niezdolność patrzenia w górę i w dół, trudności w chodzeniu i połykaniu, postępującą utratę słuchu i postępującą demencja. Ludzie z typ C mają tylko łagodne powiększenie śledziony i wątroby. Oczekiwana długość życia osób z typ C różni się znacznie. Niektóre osoby umierają w dzieciństwie, podczas gdy inne mniej dotknięte chorobą mogą również żyć w wieku dorosłym.
Obecnie nie ma lekarstwa na chorobę Niemanna-Picka. Leczenie jest wspomagające. Dzieci zwykle umierają z powodu infekcji lub postępującej utraty neurologicznej. Odnotowano przypadki przeszczepu szpiku kostnego u kilku pacjentów z: typ B z mieszanymi wynikami.
Choroba Fabry'egoznany również jako niedobór alfa-galaktozydazy-A, powoduje nagromadzenie substancji tłuszczowych w autonomicznym układzie nerwowym (tej części układu nerwowego, która kontroluje mimowolne funkcje, takie jak oddychanie i bicie serca), w oczach, nerkach i układzie krążenia system.
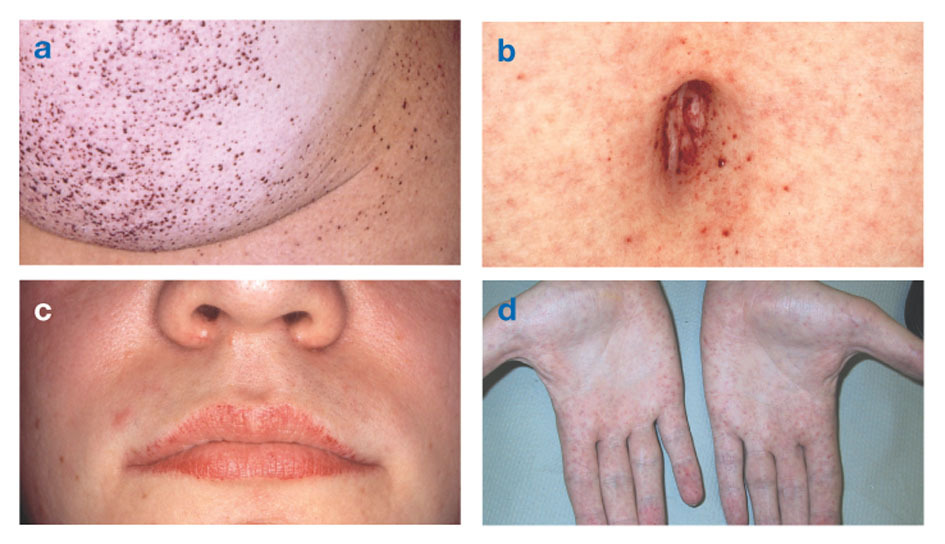
Choroba Fabry'ego jest jedyną chorobą lipidową sprzężoną z chromosomem X. Choroba dotyka głównie mężczyzn, chociaż łagodniejszą i bardziej zmienną postać występuje u kobiet. Rzadko kobiety dotknięte chorobą mają poważne objawy podobne do tych obserwowanych u mężczyzn z zaburzeniem. Początek objawów zwykle występuje w dzieciństwie lub w okresie dojrzewania.
Objawy neurologiczne obejmują piekący ból w rękach i nogach, który nasila się w czasie upałów lub później ćwiczenia, a także gromadzenie się nadmiaru materiału w przezroczystych warstwach rogówki (co prowadzi do zmętnienia, ale bez zmiany widzenia). Nagromadzenie tłuszczu w ścianach naczyń krwionośnych może zakłócić krążenie, narażając osobę na ryzyko udar mózgu lub atak serca. Inne objawy to powiększenie serca, postępujące niewydolność nerekprowadzące do schyłkowej niewydolności nerek, problemów żołądkowo-jelitowych, zmniejszonej potliwości i gorączki.
Angiokeratomy (małe, nienowotworowe, czerwonawo-fioletowe wypukłe plamy na skórze) mogą rozwijać się w dolnej części tułowia i z wiekiem stają się częstsze.
Osoby z chorobą Fabry'ego często umierają przedwcześnie z powodu powikłań spowodowanych niewydolność serca, niewydolność nerek lub udar. Leki takie jak fenytoina i karbamazepina są często przepisywane w celu leczenia bólu towarzyszącego chorobie Fabry'ego, ale nie w celu jego wyleczenia. Metoklopramid lub Lipisorb (suplement diety) mogą łagodzić zaburzenia żołądkowo-jelitowe, które często występuje u osób z chorobą Fabry'ego, a niektóre osoby mogą potrzebować przeszczepu nerki lub dializa. Zastąpienie enzymu może zmniejszyć przechowywanie, złagodzić ból i zachować funkcję narządów u niektórych osób z chorobą Fabry'ego.
choroba Farbera znany również jako Lipogranulomatoza Farbera, opisuje grupę rzadkich zaburzeń autosomalnych recesywnych, które powodują gromadzenie się tkanki tłuszczowej w stawach, tkankach i ośrodkowym układzie nerwowym. Dotyczy zarówno mężczyzn, jak i kobiet. Choroba zwykle zaczyna się we wczesnym dzieciństwie, ale może wystąpić w późniejszym życiu.
Rozwijają się dzieci z klasyczną chorobą Farbera objawy neurologiczne, które mogą obejmować zwiększoną senność i letarg oraz problemy z łykanie. Może to mieć również wpływ na wątrobę, serce i nerki. Inne objawy mogą obejmować przykurcze stawów (przewlekłe skrócenie mięśni lub ścięgien wokół stawów), wymioty, zapalenie stawów, powiększenie węzły chłonne, zapalenie stawów, chrypka i grudki pod skórą, które gęstnieją wokół stawów w miarę postępu choroby.
Osoby dotknięte chorobą z trudnościami w oddychaniu mogą wymagać włożenia rurki do oddychania. Większość dzieci z tym schorzeniem umiera w wieku 2 lat, zwykle od choroby płuc. W jednej z najcięższych postaci choroby, wkrótce po urodzeniu można zdiagnozować powiększenie wątroby i śledziony. Dzieci urodzone z tą postacią choroby zwykle umierają w ciągu 6 miesięcy.
Choroba Farbera jest spowodowana niedoborem enzymu zwanego ceramidazą. Obecnie nie ma specyficznego leczenia choroby Farbera. W celu złagodzenia bólu można przepisać kortykosteroidy. Przeszczep szpiku kostnego może zmniejszyć ziarniniaki (małe masy tkanki objętej stanem zapalnym) u osób z łagodnym stanem zapalnym lub u pacjentów bez powikłań płucnych lub układu nerwowego. U osób starszych ziarniniaki można zmniejszyć lub usunąć chirurgicznie.
Gangliozydoza składa się z dwóch różnych grup chorób genetycznych. Oba są autosomalne recesywne i w równym stopniu wpływają na mężczyzn i kobiety.
Gangliozydoza GM1.
Gangliozydoza GM1 jest spowodowana niedoborem enzymu beta-galaktozydazy, co skutkuje nieprawidłową akumulacją kwaśne materiały lipidowe, w szczególności w komórkach nerwowych w ośrodkowym i obwodowym układzie nerwowym systemy. Gangliozydoza GM1 ma trzy objawy kliniczne:
- GM1 (najcięższy podtyp, występujący wkrótce po urodzeniu) może obejmować neurodegenerację, drgawki, powiększenie wątroby i śledziony, szorstkość rysów twarzy, nieregularności szkieletu, sztywność stawów, wzdęcia, osłabienie mięśni, nadmierna reakcja na przestraszenie i problemy z chód. U około połowy osób dotkniętych chorobą pojawiają się wiśniowoczerwone plamy w oku. Dzieci mogą być głuche i niewidome w wieku 1 roku i często umierają w wieku 3 lat z powodu powikłań sercowych lub zapalenie płuc.
- Późne dziecko Gangliozydoza GM1 zwykle rozpoczyna się w wieku od 1 do 3 lat. Objawy neurologiczne obejmują ataksję, drgawki, demencję i trudności w mówieniu.
- Gangliozydoza GM1 rozwija się w wieku od 3 do 30 lat. Objawy obejmują zmniejszenie masy mięśniowej (zanik mięśni), powikłania neurologiczne, które są mniej poważne i postępują wolniej niż inne formy zaburzenia, zmętnienie rogówki u niektórych osób i utrzymujące się skurcze mięśni, które powodują skręcanie i powtarzalne ruchy lub nieprawidłową postawę (dystonia). Angiokeratomy mogą rozwijać się w dolnej części tułowia. Wielkość wątroby i śledziony jest normalna u większości osób dotkniętych chorobą.
Gangliozydoza GM2.
Gangliozydoza GM2 powoduje również, że organizm gromadzi nadmiar kwaśnych substancji tłuszczowych w tkankach i komórkach, głównie komórkach nerwowych. Zaburzenia te wynikają z niedoboru enzymu beta-heksozoaminidazy. Zaburzenia GM2 obejmują:
- Choroba Taya-Sachsa (znana również jako wariant B gangliozydozy GM2) i jej odmiany są spowodowane niedoborem enzymu heksozoaminidazy A. Dzieci dotknięte chorobą rozwijają się normalnie w ciągu pierwszych kilku miesięcy życia. Objawy zaczynają się w wieku 6 miesięcy i obejmują postępującą utratę zdolności umysłowych, otępienie, zmniejszony kontakt wzrokowy, nasilony reakcja zaskoczenia na hałas, postępująca utrata słuchu prowadząca do głuchoty, trudności w połykaniu, ślepota, wiśniowe plamy na siatkówce i niektóre paraliż. Napady mogą rozpocząć się w drugim roku życia dziecka. Niemowlęta mogą w końcu stać się zależne od zgłębnika i często umierają w wieku 4 lat z powodu nawracających infekcji. Niestety nie ma specjalnego leczenia. Leki przeciwdrgawkowe mogą początkowo kontrolować napady padaczkowe. Inne terapie wspomagające obejmują prawidłowe odżywianie i nawodnienie oraz metody utrzymywania drożności dróg oddechowych. Rzadka postać zaburzenia zwana choroba Tay-Sachsa o późnym początku, występuje u osób w wieku od 20 do 30 lat i charakteryzuje się niestabilnym chodem i postępującym pogorszeniem stanu neurologicznego.
- choroba Sandhoffa (Opcja AB) jest ciężką postacią choroby Taya-Sachsa. Początek choroby zwykle występuje w wieku 6 miesięcy i nie jest ograniczony do żadnej grupy etnicznej. Objawy neurologiczne mogą obejmować postępujące pogorszenie ośrodkowego układu nerwowego, ruchowego osłabienie, wczesna ślepota, ciężka przestraszona reakcja na dźwięk, spastyczność, wstrząs lub drżenie mięśnie (mioklonie), padaczka, nieprawidłowo powiększona głowa (makrocefalia) i czerwone plamy na oku. Inne objawy mogą obejmować częste infekcje dróg oddechowych, szmer serca, rysy twarzy przypominające lalkę oraz powiększenie wątroby i śledziony. Nie ma specyficznego leczenia choroby Sandhoffa. Podobnie jak w przypadku choroby Tay-Sachsa, leczenie podtrzymujące obejmuje utrzymywanie dróg oddechowych przez cały czas, a także prawidłowe odżywianie i nawodnienie. Leki przeciwdrgawkowe mogą początkowo kontrolować padaczkę (napady padaczkowe).
choroba Krabbego(znany również jako leukodystrofia komórek globularnych oraz lipidoza galaktozyloceramidowa) jest chorobą autosomalną recesywną spowodowaną niedoborem enzymu galaktocerebrozydazy. Choroba najczęściej dotyka niemowlęta, które zachorowały przed 6. miesiącem życia, ale może wystąpić w okresie dojrzewania lub dorosłości.
Nagromadzenie niestrawionych tłuszczów wpływa na wzrost osłonki ochronnej nerwu (osłonki mielinowej) i powoduje poważne upośledzenie zdolności umysłowych i motorycznych. Inne objawy to osłabienie mięśni, zmniejszona zdolność mięśni do rozciągania (hipotonia), napięcie mięśni (spastyczność), nagły wstrząs lub drganie kończyn (drgawki miokloniczne), drażliwość, niewyjaśniona gorączka, głuchota, ślepota, paraliż i trudności w połykaniu. Może również wystąpić długotrwała utrata wagi.
Chorobę można zdiagnozować za pomocą testów enzymatycznych i identyfikacji charakterystycznych grup komórek do ciał globoidalnych w istocie białej mózgu, demielinizacji nerwów i degeneracji oraz niszczenia komórek mózgowych. U niemowląt choroba zwykle kończy się śmiercią przed ukończeniem 2. roku życia. Osoby z bardziej zaawansowaną postacią choroby mają łagodniejszy przebieg choroby i żyją znacznie dłużej.
Nie opracowano żadnego konkretnego leczenia choroby Krabbego, chociaż wczesna transplantacja szpiku kostnego może pomóc niektórym osobom. Osoby z bardziej zaawansowaną postacią choroby mają łagodniejszy przebieg choroby i żyją znacznie dłużej.
Leukodystrofia metachromatyczna, lub MLD, to grupa zaburzeń charakteryzujących się nagromadzeniem substancji tłuszczowych w istocie białej ośrodkowego układu nerwowego oraz w nerwach obwodowych i do pewnego stopnia w nerkach. Podobnie jak choroba Krabbego, MLD wpływa na mielinę, która pokrywa i chroni nerwy. To autosomalne recesywne zaburzenie spowodowane jest niedoborem enzymu arylosulfatazy A. To zaburzenie dotyka zarówno mężczyzn, jak i kobiety.
Leukodystrofia metachromatyczna ma trzy charakterystyczne formy: późne niemowlę, młodociane i dorosłe.
- Późne niemowlę MLD zwykle zaczyna się między 12 a 20 miesiącem po urodzeniu. Dzieci mogą początkowo wyglądać normalnie, ale mają trudności z chodzeniem i skłonnością do upadków, czemu towarzyszy sporadyczny ból rąk i nóg. postępująca utrata wzroku prowadząca do ślepoty, opóźnień rozwojowych i utraty wcześniej nabytych kamieni milowych, upośledzenia połykania, drgawek i demencji do 2 lat. Dzieci rozwijają również postępujący zanik mięśni i osłabienie, a ostatecznie tracą zdolność chodzenia. Większość dzieci z tą postacią choroby umiera przed ukończeniem 5 roku życia.
- MLD dla nieletnich zwykle zaczyna się w wieku od 3 do 10 lat. Objawy obejmują upośledzenie wyników w nauce, upośledzenie umysłowe, ataksję, drgawki i demencję. Objawy postępują, a śmierć następuje po 10–20 latach od zachorowania.
- Postać dla dorosłych. Objawy dorośli ludzie początek po 16 roku życia i może obejmować ataksję, drgawki, nieprawidłowe drżenie kończyn (drżenie), zaburzenia koncentracji, depresja, zaburzenia psychiczne i demencja. Śmierć zwykle następuje po 6–14 latach od wystąpienia objawów.
MLD nie można wyleczyć. Leczenie jest objawowe i podtrzymujące. Przeszczep szpiku kostnego może w niektórych przypadkach opóźnić postęp choroby. Poczyniono znaczne postępy w terapii genowej na modelach zwierzęcych z MLD oraz w badaniach klinicznych.
choroba Wolmanaznany również jako niedobór lizosomalnej kwaśnej lipazyjest poważnym zaburzeniem spichrzania lipidów, które zwykle kończy się zgonem w wieku 1 roku. To autosomalne recesywne zaburzenie charakteryzuje się nagromadzeniem estrów cholesterolu (zwykle formy transportowej cholesterolu) i trójglicerydy (postać chemiczna, w której tłuszcze występują w organizmie), które mogą się znacznie kumulować i powodować uszkodzenia komórek oraz tekstylia.
Na to zaburzenie cierpią zarówno mężczyźni, jak i kobiety. Niemowlęta są normalne i aktywne po urodzeniu, ale szybko rozwijają się postępujące upośledzenie umysłowe, powiększona wątroba i znacznie powiększona śledziona, wzdęcia, problemy żołądkowo-jelitowe. żółtaczka, niedokrwistość, wymioty i złogi wapnia w nadnerczapowodując ich stwardnienie.
Inny typ niedobór lizosomalnej kwaśnej lipazy jest choroba magazynowania estrów cholesterolu. Ta niezwykle rzadka choroba występuje w wyniku przechowywania estrów cholesterolu i trójglicerydy w komórkach krwi, limfa i tkanka limfatyczna. U dzieci w wieku dorosłym wątroba powiększa się, co prowadzi do marskości i chroniczny niewydolność wątroby. Dzieci mogą również mieć złogi wapnia w nadnerczach, aw późniejszych stadiach choroby może rozwinąć się żółtaczka.
Obecnie temat zastępowania enzymów jest aktywnie badany zarówno w chorobie Wolmana, jak iw akumulacji estru cholesterolu.
Uwaga! Informacje służą wyłącznie celom informacyjnym i nie mają na celu diagnozowania i przepisywania leczenia. Zawsze skonsultuj się z lekarzem specjalistą!